Fusion Protein Isolation 融合蛋白分离纯化
互联网
1.Start a 5 ml overnight culture from a frozen stock (in the -70℃)in the following media:
For 100 ml of M9CA media tyrp
10 ml of 10X M9 salts
5 ml of 10% casamino acids
0.2 ml 1M MgSO4
0.01 ml 1M CaCl2
0.5 ml 40% glucose
20µl of 50 mg/ml thiamine
0.4 ml 5 mg/ml tryptophan
84 ml sterile H2O
and add ampicillin to 100μg/ml to ensure bacteria retains the plasmid
Shake culture overnight at 37℃ for 8-14 hrs.Make sure the overnight is fresh.Do not use cells that have been stored at 4℃ or have been at stationary phase for several hours.
The next day inoculate 1 ml of the overnight culture into 10mls of the same media and grow to late log phase OD599 between 0.5 to 0.8.
2.Spin down cells and resuspend into 200 ml of M9CA media as above except NO tyrptophan,and place in a sterile 1 or 2 liter flask.
3.Shake until OD599nm=0.2-0.3.This will take between 1-4 hours.
4.Add indoleacrylic acid to 30 µg/ml (600µl of a 5 mg/ml stock made fresh in ethanol).The IAA must be fresh or the induction will be poor.
5.Shake at 37℃ for 4-6 hours to induce overexpression of protein.
6.Store culture overnight in coldroom.
Next day,
7.Spin down cells in 2 centrifuge tubes.Resuspend in 2 ml of Cracking buffer.
Cracking buffer is:
6M Urea
1% SDS
10mM Na phosphate,pH 7.2 this solution can be mixed and stored as a stock.When resuspending cells add β-ME to 1%.
8.Add 20µl of β-mercaptoethanol to the resuspended cells,as mentioned above in cracking buffer recipe.
9.Incubate at 37℃ for 30 min.
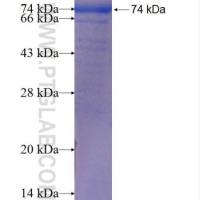
10.At this point,you are ready to load the gel for protein isolation.However,the lysate should not be too viscous that elution from a pipette is difficult,due to the DNA.If this is the case,first add additional cracking buffer (1 ml)and mix well You can also add sample buffer to follow the dye front.Add 1-1.5 ml of lysate to top of large thick gel and run (Overnight at 75 mAmps).
11.Stain running gel in 0.05% comassie blue (R-250)in water for 15-30 min.The comassie is made up as a 5% stock in MeOH and then diluted in water.
12.Destain in several washes of water for 1-2 hours until you can visualize the desired band.
Avoid using methanol and acetic acid during the stain or destain procedure since this will fix the protein into the gel and reduce the yeild during electroelution.
13.Cut out the desired overproduced band with a fresh razor blade,and chop into fine blocks of 1 mm.
14.Soak dialysis tubing (10-14 Kd cut-off)in elution buffer for 2 hours prior to use and then place over the elution chamber cell openings,both the large and small holes of the plastic container.
15.Place gel cubes in the large well of the elution cell,cover with elution buffer,and add 10% DTT to 0.1% (10µl/ml and usually ~1.5ml of elution buffer to cover the gel cubes.
16.Overlay with more buffer till both wells filled and the whole cell chamber is filled to the upper level.
17.Run overnight at 50V (12-16 hours).Black electrode on gel side to elute protein.
Next day,
18.Remove liuid carefully from the top of collection well and save till you know all protein was in the pellet that forms on the dialysis tubing.
19.With small remaining buffer over the pellet,resuspend the solid and place in a tube.Heat to get into solution.You may have to add additional SDS to get completely into solution.
20.Then run a mini-gel to quantitate the amount of protein eluted from the gel.
You can now produce antibodies with this eluted protein or make an affinity column.
Elution Buffer (make 2 liters)
0.1M Na borate (pH 8.5)Which is made as 0.1M NaOH (8g/2l)and pH with solid boric acid till pH=8.5
Add SDS to 0.01% (500µl of 20% SDS stock per 2 liters)
NOTES:
1.The trpE polypeptide portion is about 37,000 MW
2.This method has been successful in over-expressing fusion polypeptides up to 90kD.
3.To determine which band is your protein it is best done when compared to lysates of cells containing the plasmid but not induced (so you can see which band is overexpressed by induction) and also to compare with cells overexpressing just the tryE portion alone.