V294.PART II,Chapter 4 细胞迁移实验之Dunn趋化迁移培养皿法及时序显微镜分析法
丁香园
7093
PART II:BASIC CELL MIGRATION AND RELATED ASSAYS
vol.294 Guan J.-L. (ed.) Cell Migration-Developmental Methods and Protocols Chapter 3
Claire M. Wells and Anne J. Ridley
Summary
The directed migration of cells (chemotaxis) occurs not only during wound healingand inflammatory responses but also during embryonic development. However, theintracellular signaling pathways that enable a cell to detect a chemoattractant and subsequentlymigrate toward the source are not clearly defined. The Dunn chemotaxischamber in conjunction with time-lapse microscopy is a powerful tool that enables theuser to observe directly the morphological response of cells to a chemoattractant inreal time. Here, we describe using the Dunn chemotaxis chamber to study the responseof murine bone marrow-derived macrophages to colony stimulating factor-1. This is aparticularly useful protocol as it can be adapted to study bone marrow-derived macrophagesisolated from genetically modified mice and thus study the requirement for aspecific protein in cell migration and chemotaxis.
Key Words: Chemotaxis; Dunn chemotaxis chamber; cell migration; bone marrowderived macrophages; time-lapse microscopy.
1. Introduction
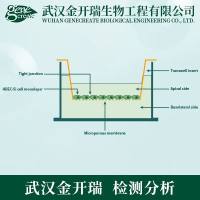
Directed cell migration (chemotaxis) is important during embryogenesis (1),wound healing (2), and inflammatory responses (3). In addition, cell migration isbelieved to contribute to the metastatic process (4). The technique described inthis chapter has been developed to study the intracellular signaling pathways andsubsequent changes in cell adhesion and morphology that are required for a cellto initiate a migratory response toward a chemoattractant. Chemotaxis can bestudied using a Boyden chamber or transwell assay; however, both these methodsare based on scoring cells that have migrated into or through a filter membranetoward a source of putative chemotactic factor. In these assays, the local concentration gradients of chemotactic factor in and around the pores of the filtermembrane are variable and unknown. Furthermore, the migratory behavior ofthe cells is unobservable and can only be deduced from the final distribution ofthe cell population. To overcome these experimental problems, the Dunnchemotaxis chamber (DCC) was developed. In this chamber, the migratorybehavior of cells can be directly observed in a gradient of known directionand magnitude. The DCC is a modification of the Zigmond chamber (5), whichallows the direct observation of slowly moving cells in a concentration gradientthat is stable over longer periods of time (6,7). The DCC consists of two concentriccircles ground into one face of a glass slide (referred to as the inner and outerwells). An annular ridge (referred to as the bridge) separates the two wells. Thebridge is 20 μm lower than the surrounding glass slide. The outer well containsthe chemoattractant whereas the inner well does not (Fig. 1). A linear gradient ofchemoattractant forms by diffusion across the bridge between these two wells.Cells seeded onto a glass cover slip are inverted over the chamber and it is thecells that lie directly above the bridge that are viewed during the assay. The DCCchamber has been successfully used to characterize the migratory response ofhuman macrophages to colony-stimulating factor-1 (CSF-1; ref. 8), fibroblaststo platelet-derived growth factor (7) and thrombospondin (TSP1) (9), neutrophilsto interleukin-8 (10), and microglia to adenosine triphosphate/adenosinediphosphate (11). In addition, the DCC has been used to study neurite outgrowth(12). We have previously used the DCC to elucidate the role of Rho family proteinsin the chemotactic response of BAC1.2F5 macrophages to CSF-1 (13). Wedescribe the use of this chamber to study the response of primary murine bonemarrow-derived macrophages (BMMs) to CSF-1, a protocol that can be extendedto study the migratory behavior of macrophages and other cell types derivedfrom genetically modified mice.
2. Materials
1. All tissue culture medium and supplements are obtained from Invitrogen unlessotherwise stated. Macrophage starve medium consists of RPMI1640 withL-glutamine, 1% essential amino acids, 1% sodium pyruvate, 1% penicillin–streptomyosin, 10% heat-inactivated fetal calf serum, and 0.5% β-mercaptoethanolsolution. The β-mercaptoethanol solution (50 mM) consists of 34 μL of14.3 M β-mercaptoethanol (Sigma; cat. no. M7522) diluted in 10 mL of RPMIcontaining antibiotics; this can be stored at 4°C. The BMMs are differentiatedand maintained in macrophage growth medium, which consists of macrophagestarve medium with the addition of 15% conditioned medium from L-929 fibroblasts(L cells) as a source of CSF-1. To provide reproducible and accurate concentrationgradients during the chemotaxis assay, recombinant CSF-1 (R and Dsystems) is used in the DCC. The BMMs are maintained in a humidified incubatorat 37°C in the presence of 10% CO2.
2. 18 × 18-mm Glass cover slips (no. 3, BDH laboratory supplies) are individuallywashed with detergent, rinsed six times with distilled water, soaked for 10 min inconcentrated HCl, then washed extensively with distilled water and stored in100% ethanol until use. Before use the ethanol is removed from each cover slipby sweeping it through a naked flame using tweezers until all the ethanol hasevaporated. This also serves to sterilize the cover slip.
3. Dunn chemotaxis chambers are supplied by Weber Scientific International. Sealingthe cover slips onto the chambers requires dental wax, which can be purchasedfrom Agar Scientific. The wax is applied to the chamber using a smallpaintbrush. Do not use a paintbrush with plastic bristles as these melt in the hotwax. We use a sable artists paintbrush.
4. It is prudent to clean the chambers immediately after use. The most importantprinciple to observe when cleaning the DCC is to avoid touching the bridge. Allthe dental wax is removed carefully with a razor blade scraping in the oppositedirection to the bridge at all times. Using gloved fingers rub off any residualwax with distilled water and rinse a further six times with distilled water. Thechamber is then placed in a glass Coplin jar (or beaker) filled with 100% acetoneand sonicated for 10 min using a water bath sonicator (Decon FS Minor, UltrasonicLtd). After sonication the chamber is rinsed six times with distilled water. Once rinsed place the chamber in a glass Petri dish grooved side upwards andsoak in 30% hydrogen peroxide for 10 min. After acid cleaning the chamber isrinsed a further six times with distilled water. Store the chambers in 100% ethanoluntil use.
5. To observe live cells in the DCC requires a microscope adapted for time-lapserecording. We use an axiovert 135 microscope (Zeiss; Hertfordshire, UK) equippedwith a heated stage (Zeiss), a Uniblitz electronic shutter (Vincent Associates;NY), and a 1CP-M1E CCD camera (Hitachi Denshi; Leeds, UK) coupled to aMeteor Frame Grabber card (Matrox Electronic Systems; Quebec, Canada). Thesystem is controlled and coordinated by Tempus meteor.exe computer software(Kinetic Imaging; Nottinghamshire, UK). Ideally, the microscope should have aheated stage, but a fan heating system can be used (14). Phase-contrast microscopyusing an inverted or upright microscope will provide good results. To determinethe chemotactic potential of a cell population (using the software describedin Subheading 3.3., step 7) requires the analysis of a large number of cells.
Therefore, a low-magnification objective lens (routinely 10X) is used wherebythe full width of the DCC bridge can be visualized.
3. Methods
3.1. Isolation and Culture of Mouse Primary BMMs1. Isolate and clean the murine femoral bone. Maintain the bones in a small Petridish containing macrophage starve medium. It is important that all of the tissuesurrounding the femur is scraped away to prevent contamination of the BMMpreparation.
2. Once the bone has been cleaned use a 19-gage needle to pierce both ends of thebone. Keep the needle in one end of the bone (fatter end). Using a 5-mL syringe,flush the bone marrow out of the bone with 5 mL of macrophage starve mediuminto a 15-mL Falcon tube. Add an additional 5 mL of macrophage starve mediumand resuspend vigorously using a p1000 Gilson pipet.
3. Centrifuge the cell suspension for 5 min at 1000g and resuspend the pellet in 5 mL of macrophage starve medium. Count the hematopoietic progenitor cells(do not count the smaller crescent-shaped red blood cells).
4. Seed the cells at 2 × 105 cells/cm2 on tissue culture plastic (Nunc). We routinelyseed 15 mL of cells suspended in growth medium in a 75-cm2 flask at a density of1 × 106 cells/mL. Incubate the cells for 3 d.
5. Carefully collect the non-adherent population of cells. The cells attached to thebottom of the flask are a mixed population of differentiated hematopoietic cellsand fibroblasts and care should be taken not to dislodge these cells when collectingthe non-adherent population. Centrifuge the cell suspension and count all thecells present.
6. The cells are then cryogenically frozen in growth medium containing 10% DMSOat a cell density depending on the subsequent application. We routinely placemultiples of 5 × 105 cells in each vial.
7. To recover frozen cells rapidly thaw a cryovial, add 4 mL of warm growthmedium dropwise and centrifuge for 5 min at 1000g.
8. Seed the cells on 6-cm bacterial culture plates (Falcon; see Note 1) in 5 mL ofgrowth medium at a density of 105 cells/mL (i.e., a vial containing 5 × 105 cells will seed one 6-cm dish). One 6-cm dish will produce enough cells for over 20Dunn chamber cover slips.
9. Incubate the cells for a further 5 d without refeeding. The cells remain in suspensionuntil approximately d 4. The differentiated BMMs will then become adherentand can be harvested on d 5 (see Note 1).
10. To harvest the cell carefully remove the medium as the cells are only looselyattached and add 2.5 mL of Versene (Gibco). Incubate for approx 20 min andthen collect the cells suspension by vigorous pipetting across the dish. Add 2.5mL of macrophage starve medium and centrifuge for 5 min at 1000g. Resuspendthe cells in an appropriate amount of macrophage growth medium.
11. Place a flamed (see Subheading 2, item 2) acid-washed Dunn chamber coverslip in a 2-cm tissue culture dish and add 2 mL of growth medium containing 2 ×104 cells/mL. Incubate the cells for a further 18 h.
12. To prepare the cells for the DCC, wash twice with starve medium and then incubatein starve medium for 8 h. At the same time place 8 mL of starve mediumin the incubator, so that it becomes warm and gassed, to use in the chamberassembly.
3.2. Assembly of the DCC
1. Place some dental wax in a glass beaker and melt slowly using a low setting ona stage heater. Avoid overheating the wax, as a noxious fume will be produced.
Ensure that the time-lapse stage is heated to 37°C and that filming can beginimmediately after the chamber is assembled.
2. Aliquot 5 mL of the pregassed (see Subheading 3.1., step 12) starve mediuminto a 7-mL bijoux tube: this will be used to wash the chambers (wash). Aliquot1 mL of the starve medium into a 7-mL bijoux tube (control) and 1 mL of starvemedium into a 7-mL bijoux tube (test). Add 30 ng/mL recombinant CSF-1 to the1 mL of starve medium in the test bijoux tube.
3. Place the cover slip with cells attached, wash, control and test bijoux tubes in anincubator close to the DCC assembly site. Alternatively 5 or 10% CO2 can bepumped from a cylinder cross the DCC assembly site. It is important to keep themedium used during assembly of the DCC fully gassed, as once the chamber issealed by wax there is no opportunity for gas exchange.
4. Place a DCC on tissue paper until the storage ethanol has evaporated. The chambershould be completely dry before proceeding. Do not attempt to wipe the chamberas contact with the bridge can result in damage that may affect the gradient.
5. Initially the chamber is washed to remove any residual traces of ethanol. Using aGilson, carefully pipet 100–150 μL of wash medium over the center of the chamberwithout letting the tip touch the bridge. Both wells should fill but not overflow. Carefully remove the medium and repeat six times.
6. Place 150 μL of control medium over the chamber until it fills both wells. Quicklybut carefully invert the cover slip over the two wells in the center of the chamber. It is important that inverting the cover slip over the chamber does not incorporateany bubbles into the chamber as these can affect the gradient (see Note 2). The cover slip should be placed slightly off center so that a small gap remains inthe outer well (Fig. 2). However, the cover slip should completely cover theinner well so that the outer well can be drained without any loss of medium fromthe inner well.
7. Lightly press down the cover slip around the edges and mop up excess mediumwith Whatman paper. Using a sable paintbrush wax three sides of the cover slipleaving the side with the outer well gap unwaxed (Fig. 2).
8. Tear a piece of Whatman paper and place a small corner of the torn edge justinside the outer well gap until it starts to absorb the medium. Leave the filterpaper to absorb all of the medium in the outer well. It is important not to move orlift the paper as this can introduce air into the DCC.
9. Using a Gilson pipet add 100 μL of test solution into the outer well through thegap, making sure that it is bubble-free, until the well is full (see Note 3 for testingthe gradient formation). Quickly wax the remaining side ensuring that the gap iscompletely sealed. Once the wax has set wash the remaining cover slip surfacewith distilled water and dry with Whatman paper or compressed air.
10. Immediately place the DCC on the microscope stage and start filming. See Note 4for fixing cells at the end of filming.
3.3. Time-Lapse Microscopy and Migration Analysis
1. Place the assembled chamber on the heated stage.
2. The live image from the microscope is then viewed on the computer monitorusing the Acquisition manager (AQM) software (Kinetic Imaging; Liverpool,UK) and a region of the bridge is selected for recording. Ideally, there should be15–25 cells in the field of view and the cells should be evenly spaced.
3. The high concentration (outer well) end of the chemotactic gradient is thenaligned to the top of the monitor screen (Fig. 3). We achieve this by rotating thecamera. This is extremely useful for subsequent tracking and mathematical analysisof chemotactic behavior.
4. For analyzing the chemotactic behavior of BMMs in a gradient of recombinantCSF-1, we use a time-lapse interval of 10 min and film the cells for 24 h. BMMsrespond slowly to a CSF-1 gradient compared with macrophage cell lines such asBac1.2F5 (13).
5. Once the assay is finished, the recorded sequence of images can be analyzedusing the same AQM software (Kinetic Imaging). Each cell must be individuallytracked using the AQM software throughout the sequence. In our laboratory, weadopt the following tracking criteria: only cells present in the first frame aretracked. If any cell present in the first frame divides within the first 60 frames, itis excluded. If a cell present in the first frame divides at a later stage in the film,it is tracked until it ceases to be migratory; for BMMs this is normally one to twoframes or 20 min before mitosis occurs. The daughter cells of this division arenot tracked.
6. Once all the appropriate cells in the field of view have been tracked the markedpositions of each cell in each frame are saved into a file with the extension “.cel.”
This file records the cell number, frame number, x-coordinate and y-coordinateof every tracked cell in every frame.
7. Finally, we conduct trajectory analysis using a range of software originallydesigned by Dr. Graham Dunn and Dr. Daniel Zicha at the Randall Centre, KingsCollege London. This program returns a Rayleigh test for unimodal clusteringof directions where the null hypothesis is uniform distribution of cell trajectories.
If the null hypothesis is rejected by the Rayleigh test the cells are showinga significant directional response to CSF-1. This software is not commerciallyavailable, however Kinetic Imaging has some software analysis to use with theDCC incorporated into the AQM software.
8. The cell trajectories can be plotted so that their starting point is shifted to theorigin in Microsoft Excel to give a preliminary indication of chemotaxis. A significantchemotactic response should result in the majority of cell trajectoriesradiating from the origin in the direction of the highest chemoattractant concentration(ref. 10 and Fig. 4). To do this the tracking data must also be saved asan .xld file, which can be recognized by Excel. This option is available in theKinetic Imaging tracking software.
4. Notes
1. The BMMs must be differentiated and cultured on bacterial plates because theyadhere too strongly to tissue culture plastic and are impossible to passage. BMMschange in morphology and migratory behavior during passaging. They graduallybecome more elongated and less motile thus reducing their chemotactic potential.
In our motility assays, we only use cells for 7 d after the 5-d differentiationperiod.
2. Successful assembly of the DCC is a specialized technique that requires patienceand practice. It is advisable for a new user to practice assembling the chamberinitially using a wet cover slip with no cells attached. It is essential that the usercan assemble a chamber that contains no bubbles after a 24-h incubation. Thepresence of bubbles particularly in the inner well will seriously interfere withthe formation of the gradient and produce unreliable results. Bubbles can resultfrom incorrect sealing of the chamber but are most commonly the result of thecover slip being lifted away from the chamber during the draining of the outerwell. Once the chamber can be assembled correctly the user should be able toproceed to assembling the chamber with cover slips seeded with cells. If thecells are still viable 24 h later then the user can confidently begin to conductchemo-taxis assays.
3. The time taken for a gradient to form between the two wells of the DCC dependson the molecular weight of the chemotactic factor. Factors with a molecularweight between 350 and 370 Da will form a gradient in 10 min, whereas factorswith a molecular weight between 10 and 20,000 Da will take 30 min (7). Similarly,the molecular weight of the chemoattractant will determine the stability of the gradient—generally the greater the molecular weight the longer the gradientwill be maintained. It is advisable for a new user to test the formation and stabilityof the gradient in the DCC using a fluorescein isothiocyanate-conjugated dextranof comparable molecular weight to the intended chemoattractant. Assemblethe chamber as normal using the fluorescein isothiocyanate–dextran in the outer well. The formation of the gradient can then be monitored using a standard epifluorescencemicroscope. A similar method was used to establish that a gradientof CSF-1 forms within 25 min and is stable for at least 17 h in the DCC (15).
4. In addition to studying the response of BMMs isolated from knock-out mice, theDCC can be used to characterize the chemotactic response of cells expressingtagged proteins introduced either by microinjection of recombinant protein ordeoxyribonucleic acid (16) or by transfection. To identify expressing cells thecover slip is removed from the chamber and the cells fixed immediately after thefilming period ends. To fix the cells, remove the wax using a razor blade andcarefully lift the cover slip away from the chamber using watchmaker forceps. Immediately place the cover slip in fixative (e.g., 4% paraformaldehyde). Thecover slip can then be stained for expressed protein following normal protocols.If a GFP tag is used in conjunction with time-lapse fluorescence microscopy thecells can be directly identified and tracked from the resulting film.
References
1. Dormann, D. and Weijer, C. J. (2003) Chemotactic cell movement during development.Curr. Opin. Genet. Dev. 13, 358–364.
2. Seppa, H., Grotendorst, G., Seppa, S., Schiffmann, E., and Martin, G. R. (1982)Platelet-derived growth factor is chemotactic for fibroblasts. J. Cell Biol. 92,584–588.
3. Jones, G. E. (2000) Cellular signaling in macrophage migration and chemotaxis.
J. Leukoc. Biol. 68, 593–602.
4. Kassis, J., Lauffenburger, D. A., Turner, T., and Wells, A. (2001) Tumor invasionas dysregulated cell motility. Semin. Cancer Biol. 11, 105–117.
5. Zigmond, S. H. and Hirsch, J. G. (1973) Leukocyte locomotion and chemotaxis.New methods for evaluation, and demonstration of a cell-derived chemotacticfactor. J. Exp. Med. 137, 387–410.
6. Dunn, G. A. and Zicha, D. (1993) Long-term chemotaxis of neutrophils in stablegradients: preliminary evidence of periodic behavior. Blood Cells 19, 25–39; discussion39–41.
7. Zicha, D., Dunn, G. A., and Brown, A. F. (1991) A new direct-viewing chemotaxischamber. J. Cell Sci. 99, 769–775.
8. Jones, G. E., Zicha, D., Dunn, G. A., Blundell, M., and Thrasher, A. (2002) Restorationof podosomes and chemotaxis in Wiskott-Aldrich syndrome macrophagesfollowing induced expression of WASp. Int. J. Biochem. Cell Biol. 34,806–815.
9. Orr, A. W., Elzie, C. A., Kucik, D. F., and Murphy-Ullrich, J. E. (2003)Thrombospondin signaling through the calreticulin/LDL receptor-related proteinco-complex stimulates random and directed cell migration. J. Cell Sci. 116,2917–2927.
10. Zicha, D., Allen, W. E., Brickell, P. M., Kinnon, C., Dunn, G. A., Jones, G. E.,et al. (1998) Chemotaxis of macrophages is abolished in the Wiskott-Aldrichsyndrome. Br. J. Haematol. 101, 659–665.
11. Honda, S., Sasaki, Y., Oshawa, K., Imai, Y., Nakamura, Y., Inoue, K., andKohsaka, S. (2001) Extracellular ATP or ADP induce chemotaxis of culturedmicroglia through Gi/o-coupled P2Y receptors. J. Neurosci. 21, 1975–1982.
12. Maden, M., Keen, G., and Jones, G. E. (1998) Retinoic acid as a chemotacticmolecule in neuronal development. Int. J. Dev. Neurosci. 16, 317–322.
13. Allen, W. E., Zicha, D., Ridley, A. J., and Jones, G. E. (1998) A role for Cdc42 inmacrophage chemotaxis. J. Cell Biol. 141, 1147–1157.
14. Jones, G. E., Ridley, A. J., and Zicha, D. (2000) Rho GTPases and cell migration:measurement of macrophage chemotaxis. Methods Enzymol. 325, 449–462.
15. Webb, S. E., Pollard, J. W., and Jones, G. E. (1996) Direct observation and quantificationof macrophage chemoattraction to the growth factor CSF-1. J. CellSci.109, 793–803.
16. Ridley, A. J. (1998) Mammalian cell microinjection assay to study the function ofRac and Rho. Methods Mol. Biol. 84, 153–160.
vol.294 Guan J.-L. (ed.) Cell Migration-Developmental Methods and Protocols Chapter 3
Analysis of Cell Migration Using the Dunn Chemotaxis Chamber and Time-Lapse Microscopy
Claire M. Wells and Anne J. Ridley
Summary
The directed migration of cells (chemotaxis) occurs not only during wound healingand inflammatory responses but also during embryonic development. However, theintracellular signaling pathways that enable a cell to detect a chemoattractant and subsequentlymigrate toward the source are not clearly defined. The Dunn chemotaxischamber in conjunction with time-lapse microscopy is a powerful tool that enables theuser to observe directly the morphological response of cells to a chemoattractant inreal time. Here, we describe using the Dunn chemotaxis chamber to study the responseof murine bone marrow-derived macrophages to colony stimulating factor-1. This is aparticularly useful protocol as it can be adapted to study bone marrow-derived macrophagesisolated from genetically modified mice and thus study the requirement for aspecific protein in cell migration and chemotaxis.
Key Words: Chemotaxis; Dunn chemotaxis chamber; cell migration; bone marrowderived macrophages; time-lapse microscopy.
1. Introduction
Directed cell migration (chemotaxis) is important during embryogenesis (1),wound healing (2), and inflammatory responses (3). In addition, cell migration isbelieved to contribute to the metastatic process (4). The technique described inthis chapter has been developed to study the intracellular signaling pathways andsubsequent changes in cell adhesion and morphology that are required for a cellto initiate a migratory response toward a chemoattractant. Chemotaxis can bestudied using a Boyden chamber or transwell assay; however, both these methodsare based on scoring cells that have migrated into or through a filter membranetoward a source of putative chemotactic factor. In these assays, the local concentration gradients of chemotactic factor in and around the pores of the filtermembrane are variable and unknown. Furthermore, the migratory behavior ofthe cells is unobservable and can only be deduced from the final distribution ofthe cell population. To overcome these experimental problems, the Dunnchemotaxis chamber (DCC) was developed. In this chamber, the migratorybehavior of cells can be directly observed in a gradient of known directionand magnitude. The DCC is a modification of the Zigmond chamber (5), whichallows the direct observation of slowly moving cells in a concentration gradientthat is stable over longer periods of time (6,7). The DCC consists of two concentriccircles ground into one face of a glass slide (referred to as the inner and outerwells). An annular ridge (referred to as the bridge) separates the two wells. Thebridge is 20 μm lower than the surrounding glass slide. The outer well containsthe chemoattractant whereas the inner well does not (Fig. 1). A linear gradient ofchemoattractant forms by diffusion across the bridge between these two wells.Cells seeded onto a glass cover slip are inverted over the chamber and it is thecells that lie directly above the bridge that are viewed during the assay. The DCCchamber has been successfully used to characterize the migratory response ofhuman macrophages to colony-stimulating factor-1 (CSF-1; ref. 8), fibroblaststo platelet-derived growth factor (7) and thrombospondin (TSP1) (9), neutrophilsto interleukin-8 (10), and microglia to adenosine triphosphate/adenosinediphosphate (11). In addition, the DCC has been used to study neurite outgrowth(12). We have previously used the DCC to elucidate the role of Rho family proteinsin the chemotactic response of BAC1.2F5 macrophages to CSF-1 (13). Wedescribe the use of this chamber to study the response of primary murine bonemarrow-derived macrophages (BMMs) to CSF-1, a protocol that can be extendedto study the migratory behavior of macrophages and other cell types derivedfrom genetically modified mice.
Fig. 1. The Dunn chemotaxis chamber with cover slip in place. The chamber consistsof two concentric circles ground into one face of a glass slide (inner and outerwells). An annular ridge (bridge) separates the two wells. Insert, the bridge is 20 μmlower than the surrounding glass slide resulting in a gap between the cover slip and thebridge. Adapted from a figure kindly provided by Professor Gareth Jones.
2. Materials
1. All tissue culture medium and supplements are obtained from Invitrogen unlessotherwise stated. Macrophage starve medium consists of RPMI1640 withL-glutamine, 1% essential amino acids, 1% sodium pyruvate, 1% penicillin–streptomyosin, 10% heat-inactivated fetal calf serum, and 0.5% β-mercaptoethanolsolution. The β-mercaptoethanol solution (50 mM) consists of 34 μL of14.3 M β-mercaptoethanol (Sigma; cat. no. M7522) diluted in 10 mL of RPMIcontaining antibiotics; this can be stored at 4°C. The BMMs are differentiatedand maintained in macrophage growth medium, which consists of macrophagestarve medium with the addition of 15% conditioned medium from L-929 fibroblasts(L cells) as a source of CSF-1. To provide reproducible and accurate concentrationgradients during the chemotaxis assay, recombinant CSF-1 (R and Dsystems) is used in the DCC. The BMMs are maintained in a humidified incubatorat 37°C in the presence of 10% CO2.
2. 18 × 18-mm Glass cover slips (no. 3, BDH laboratory supplies) are individuallywashed with detergent, rinsed six times with distilled water, soaked for 10 min inconcentrated HCl, then washed extensively with distilled water and stored in100% ethanol until use. Before use the ethanol is removed from each cover slipby sweeping it through a naked flame using tweezers until all the ethanol hasevaporated. This also serves to sterilize the cover slip.
3. Dunn chemotaxis chambers are supplied by Weber Scientific International. Sealingthe cover slips onto the chambers requires dental wax, which can be purchasedfrom Agar Scientific. The wax is applied to the chamber using a smallpaintbrush. Do not use a paintbrush with plastic bristles as these melt in the hotwax. We use a sable artists paintbrush.
4. It is prudent to clean the chambers immediately after use. The most importantprinciple to observe when cleaning the DCC is to avoid touching the bridge. Allthe dental wax is removed carefully with a razor blade scraping in the oppositedirection to the bridge at all times. Using gloved fingers rub off any residualwax with distilled water and rinse a further six times with distilled water. Thechamber is then placed in a glass Coplin jar (or beaker) filled with 100% acetoneand sonicated for 10 min using a water bath sonicator (Decon FS Minor, UltrasonicLtd). After sonication the chamber is rinsed six times with distilled water. Once rinsed place the chamber in a glass Petri dish grooved side upwards andsoak in 30% hydrogen peroxide for 10 min. After acid cleaning the chamber isrinsed a further six times with distilled water. Store the chambers in 100% ethanoluntil use.
5. To observe live cells in the DCC requires a microscope adapted for time-lapserecording. We use an axiovert 135 microscope (Zeiss; Hertfordshire, UK) equippedwith a heated stage (Zeiss), a Uniblitz electronic shutter (Vincent Associates;NY), and a 1CP-M1E CCD camera (Hitachi Denshi; Leeds, UK) coupled to aMeteor Frame Grabber card (Matrox Electronic Systems; Quebec, Canada). Thesystem is controlled and coordinated by Tempus meteor.exe computer software(Kinetic Imaging; Nottinghamshire, UK). Ideally, the microscope should have aheated stage, but a fan heating system can be used (14). Phase-contrast microscopyusing an inverted or upright microscope will provide good results. To determinethe chemotactic potential of a cell population (using the software describedin Subheading 3.3., step 7) requires the analysis of a large number of cells.
Therefore, a low-magnification objective lens (routinely 10X) is used wherebythe full width of the DCC bridge can be visualized.
3. Methods
3.1. Isolation and Culture of Mouse Primary BMMs1. Isolate and clean the murine femoral bone. Maintain the bones in a small Petridish containing macrophage starve medium. It is important that all of the tissuesurrounding the femur is scraped away to prevent contamination of the BMMpreparation.
2. Once the bone has been cleaned use a 19-gage needle to pierce both ends of thebone. Keep the needle in one end of the bone (fatter end). Using a 5-mL syringe,flush the bone marrow out of the bone with 5 mL of macrophage starve mediuminto a 15-mL Falcon tube. Add an additional 5 mL of macrophage starve mediumand resuspend vigorously using a p1000 Gilson pipet.
3. Centrifuge the cell suspension for 5 min at 1000g and resuspend the pellet in 5 mL of macrophage starve medium. Count the hematopoietic progenitor cells(do not count the smaller crescent-shaped red blood cells).
4. Seed the cells at 2 × 105 cells/cm2 on tissue culture plastic (Nunc). We routinelyseed 15 mL of cells suspended in growth medium in a 75-cm2 flask at a density of1 × 106 cells/mL. Incubate the cells for 3 d.
5. Carefully collect the non-adherent population of cells. The cells attached to thebottom of the flask are a mixed population of differentiated hematopoietic cellsand fibroblasts and care should be taken not to dislodge these cells when collectingthe non-adherent population. Centrifuge the cell suspension and count all thecells present.
6. The cells are then cryogenically frozen in growth medium containing 10% DMSOat a cell density depending on the subsequent application. We routinely placemultiples of 5 × 105 cells in each vial.
7. To recover frozen cells rapidly thaw a cryovial, add 4 mL of warm growthmedium dropwise and centrifuge for 5 min at 1000g.
8. Seed the cells on 6-cm bacterial culture plates (Falcon; see Note 1) in 5 mL ofgrowth medium at a density of 105 cells/mL (i.e., a vial containing 5 × 105 cells will seed one 6-cm dish). One 6-cm dish will produce enough cells for over 20Dunn chamber cover slips.
9. Incubate the cells for a further 5 d without refeeding. The cells remain in suspensionuntil approximately d 4. The differentiated BMMs will then become adherentand can be harvested on d 5 (see Note 1).
10. To harvest the cell carefully remove the medium as the cells are only looselyattached and add 2.5 mL of Versene (Gibco). Incubate for approx 20 min andthen collect the cells suspension by vigorous pipetting across the dish. Add 2.5mL of macrophage starve medium and centrifuge for 5 min at 1000g. Resuspendthe cells in an appropriate amount of macrophage growth medium.
11. Place a flamed (see Subheading 2, item 2) acid-washed Dunn chamber coverslip in a 2-cm tissue culture dish and add 2 mL of growth medium containing 2 ×104 cells/mL. Incubate the cells for a further 18 h.
12. To prepare the cells for the DCC, wash twice with starve medium and then incubatein starve medium for 8 h. At the same time place 8 mL of starve mediumin the incubator, so that it becomes warm and gassed, to use in the chamberassembly.
3.2. Assembly of the DCC
1. Place some dental wax in a glass beaker and melt slowly using a low setting ona stage heater. Avoid overheating the wax, as a noxious fume will be produced.
Ensure that the time-lapse stage is heated to 37°C and that filming can beginimmediately after the chamber is assembled.
2. Aliquot 5 mL of the pregassed (see Subheading 3.1., step 12) starve mediuminto a 7-mL bijoux tube: this will be used to wash the chambers (wash). Aliquot1 mL of the starve medium into a 7-mL bijoux tube (control) and 1 mL of starvemedium into a 7-mL bijoux tube (test). Add 30 ng/mL recombinant CSF-1 to the1 mL of starve medium in the test bijoux tube.
3. Place the cover slip with cells attached, wash, control and test bijoux tubes in anincubator close to the DCC assembly site. Alternatively 5 or 10% CO2 can bepumped from a cylinder cross the DCC assembly site. It is important to keep themedium used during assembly of the DCC fully gassed, as once the chamber issealed by wax there is no opportunity for gas exchange.
4. Place a DCC on tissue paper until the storage ethanol has evaporated. The chambershould be completely dry before proceeding. Do not attempt to wipe the chamberas contact with the bridge can result in damage that may affect the gradient.
5. Initially the chamber is washed to remove any residual traces of ethanol. Using aGilson, carefully pipet 100–150 μL of wash medium over the center of the chamberwithout letting the tip touch the bridge. Both wells should fill but not overflow. Carefully remove the medium and repeat six times.
6. Place 150 μL of control medium over the chamber until it fills both wells. Quicklybut carefully invert the cover slip over the two wells in the center of the chamber. It is important that inverting the cover slip over the chamber does not incorporateany bubbles into the chamber as these can affect the gradient (see Note 2). The cover slip should be placed slightly off center so that a small gap remains inthe outer well (Fig. 2). However, the cover slip should completely cover theinner well so that the outer well can be drained without any loss of medium fromthe inner well.
7. Lightly press down the cover slip around the edges and mop up excess mediumwith Whatman paper. Using a sable paintbrush wax three sides of the cover slipleaving the side with the outer well gap unwaxed (Fig. 2).
8. Tear a piece of Whatman paper and place a small corner of the torn edge justinside the outer well gap until it starts to absorb the medium. Leave the filterpaper to absorb all of the medium in the outer well. It is important not to move orlift the paper as this can introduce air into the DCC.
9. Using a Gilson pipet add 100 μL of test solution into the outer well through thegap, making sure that it is bubble-free, until the well is full (see Note 3 for testingthe gradient formation). Quickly wax the remaining side ensuring that the gap iscompletely sealed. Once the wax has set wash the remaining cover slip surfacewith distilled water and dry with Whatman paper or compressed air.
10. Immediately place the DCC on the microscope stage and start filming. See Note 4for fixing cells at the end of filming.
3.3. Time-Lapse Microscopy and Migration Analysis
1. Place the assembled chamber on the heated stage.
2. The live image from the microscope is then viewed on the computer monitorusing the Acquisition manager (AQM) software (Kinetic Imaging; Liverpool,UK) and a region of the bridge is selected for recording. Ideally, there should be15–25 cells in the field of view and the cells should be evenly spaced.
3. The high concentration (outer well) end of the chemotactic gradient is thenaligned to the top of the monitor screen (Fig. 3). We achieve this by rotating thecamera. This is extremely useful for subsequent tracking and mathematical analysisof chemotactic behavior.
4. For analyzing the chemotactic behavior of BMMs in a gradient of recombinantCSF-1, we use a time-lapse interval of 10 min and film the cells for 24 h. BMMsrespond slowly to a CSF-1 gradient compared with macrophage cell lines such asBac1.2F5 (13).
5. Once the assay is finished, the recorded sequence of images can be analyzedusing the same AQM software (Kinetic Imaging). Each cell must be individuallytracked using the AQM software throughout the sequence. In our laboratory, weadopt the following tracking criteria: only cells present in the first frame aretracked. If any cell present in the first frame divides within the first 60 frames, itis excluded. If a cell present in the first frame divides at a later stage in the film,it is tracked until it ceases to be migratory; for BMMs this is normally one to twoframes or 20 min before mitosis occurs. The daughter cells of this division arenot tracked.
Fig. 3. BMMs in the DCC as imaged by AQM software. A region of the bridge isselected for recording and the image orientated such that the source of chemoattractant(outer well) is at the top of the screen.
6. Once all the appropriate cells in the field of view have been tracked the markedpositions of each cell in each frame are saved into a file with the extension “.cel.”
This file records the cell number, frame number, x-coordinate and y-coordinateof every tracked cell in every frame.
7. Finally, we conduct trajectory analysis using a range of software originallydesigned by Dr. Graham Dunn and Dr. Daniel Zicha at the Randall Centre, KingsCollege London. This program returns a Rayleigh test for unimodal clusteringof directions where the null hypothesis is uniform distribution of cell trajectories.
If the null hypothesis is rejected by the Rayleigh test the cells are showinga significant directional response to CSF-1. This software is not commerciallyavailable, however Kinetic Imaging has some software analysis to use with theDCC incorporated into the AQM software.
8. The cell trajectories can be plotted so that their starting point is shifted to theorigin in Microsoft Excel to give a preliminary indication of chemotaxis. A significantchemotactic response should result in the majority of cell trajectoriesradiating from the origin in the direction of the highest chemoattractant concentration(ref. 10 and Fig. 4). To do this the tracking data must also be saved asan .xld file, which can be recognized by Excel. This option is available in theKinetic Imaging tracking software.
Fig. 4. Trajectory plot of cells. A plot of 10 BMM cell trajectories (pixel coordinates)where their starting point is shifted to the origin in Microsoft Excel. The sourceof chemoattractant is at the top of the plot.
4. Notes
1. The BMMs must be differentiated and cultured on bacterial plates because theyadhere too strongly to tissue culture plastic and are impossible to passage. BMMschange in morphology and migratory behavior during passaging. They graduallybecome more elongated and less motile thus reducing their chemotactic potential.
In our motility assays, we only use cells for 7 d after the 5-d differentiationperiod.
2. Successful assembly of the DCC is a specialized technique that requires patienceand practice. It is advisable for a new user to practice assembling the chamberinitially using a wet cover slip with no cells attached. It is essential that the usercan assemble a chamber that contains no bubbles after a 24-h incubation. Thepresence of bubbles particularly in the inner well will seriously interfere withthe formation of the gradient and produce unreliable results. Bubbles can resultfrom incorrect sealing of the chamber but are most commonly the result of thecover slip being lifted away from the chamber during the draining of the outerwell. Once the chamber can be assembled correctly the user should be able toproceed to assembling the chamber with cover slips seeded with cells. If thecells are still viable 24 h later then the user can confidently begin to conductchemo-taxis assays.
3. The time taken for a gradient to form between the two wells of the DCC dependson the molecular weight of the chemotactic factor. Factors with a molecularweight between 350 and 370 Da will form a gradient in 10 min, whereas factorswith a molecular weight between 10 and 20,000 Da will take 30 min (7). Similarly,the molecular weight of the chemoattractant will determine the stability of the gradient—generally the greater the molecular weight the longer the gradientwill be maintained. It is advisable for a new user to test the formation and stabilityof the gradient in the DCC using a fluorescein isothiocyanate-conjugated dextranof comparable molecular weight to the intended chemoattractant. Assemblethe chamber as normal using the fluorescein isothiocyanate–dextran in the outer well. The formation of the gradient can then be monitored using a standard epifluorescencemicroscope. A similar method was used to establish that a gradientof CSF-1 forms within 25 min and is stable for at least 17 h in the DCC (15).
4. In addition to studying the response of BMMs isolated from knock-out mice, theDCC can be used to characterize the chemotactic response of cells expressingtagged proteins introduced either by microinjection of recombinant protein ordeoxyribonucleic acid (16) or by transfection. To identify expressing cells thecover slip is removed from the chamber and the cells fixed immediately after thefilming period ends. To fix the cells, remove the wax using a razor blade andcarefully lift the cover slip away from the chamber using watchmaker forceps. Immediately place the cover slip in fixative (e.g., 4% paraformaldehyde). Thecover slip can then be stained for expressed protein following normal protocols.If a GFP tag is used in conjunction with time-lapse fluorescence microscopy thecells can be directly identified and tracked from the resulting film.
References
1. Dormann, D. and Weijer, C. J. (2003) Chemotactic cell movement during development.Curr. Opin. Genet. Dev. 13, 358–364.
2. Seppa, H., Grotendorst, G., Seppa, S., Schiffmann, E., and Martin, G. R. (1982)Platelet-derived growth factor is chemotactic for fibroblasts. J. Cell Biol. 92,584–588.
3. Jones, G. E. (2000) Cellular signaling in macrophage migration and chemotaxis.
J. Leukoc. Biol. 68, 593–602.
4. Kassis, J., Lauffenburger, D. A., Turner, T., and Wells, A. (2001) Tumor invasionas dysregulated cell motility. Semin. Cancer Biol. 11, 105–117.
5. Zigmond, S. H. and Hirsch, J. G. (1973) Leukocyte locomotion and chemotaxis.New methods for evaluation, and demonstration of a cell-derived chemotacticfactor. J. Exp. Med. 137, 387–410.
6. Dunn, G. A. and Zicha, D. (1993) Long-term chemotaxis of neutrophils in stablegradients: preliminary evidence of periodic behavior. Blood Cells 19, 25–39; discussion39–41.
7. Zicha, D., Dunn, G. A., and Brown, A. F. (1991) A new direct-viewing chemotaxischamber. J. Cell Sci. 99, 769–775.
8. Jones, G. E., Zicha, D., Dunn, G. A., Blundell, M., and Thrasher, A. (2002) Restorationof podosomes and chemotaxis in Wiskott-Aldrich syndrome macrophagesfollowing induced expression of WASp. Int. J. Biochem. Cell Biol. 34,806–815.
9. Orr, A. W., Elzie, C. A., Kucik, D. F., and Murphy-Ullrich, J. E. (2003)Thrombospondin signaling through the calreticulin/LDL receptor-related proteinco-complex stimulates random and directed cell migration. J. Cell Sci. 116,2917–2927.
10. Zicha, D., Allen, W. E., Brickell, P. M., Kinnon, C., Dunn, G. A., Jones, G. E.,et al. (1998) Chemotaxis of macrophages is abolished in the Wiskott-Aldrichsyndrome. Br. J. Haematol. 101, 659–665.
11. Honda, S., Sasaki, Y., Oshawa, K., Imai, Y., Nakamura, Y., Inoue, K., andKohsaka, S. (2001) Extracellular ATP or ADP induce chemotaxis of culturedmicroglia through Gi/o-coupled P2Y receptors. J. Neurosci. 21, 1975–1982.
12. Maden, M., Keen, G., and Jones, G. E. (1998) Retinoic acid as a chemotacticmolecule in neuronal development. Int. J. Dev. Neurosci. 16, 317–322.
13. Allen, W. E., Zicha, D., Ridley, A. J., and Jones, G. E. (1998) A role for Cdc42 inmacrophage chemotaxis. J. Cell Biol. 141, 1147–1157.
14. Jones, G. E., Ridley, A. J., and Zicha, D. (2000) Rho GTPases and cell migration:measurement of macrophage chemotaxis. Methods Enzymol. 325, 449–462.
15. Webb, S. E., Pollard, J. W., and Jones, G. E. (1996) Direct observation and quantificationof macrophage chemoattraction to the growth factor CSF-1. J. CellSci.109, 793–803.
16. Ridley, A. J. (1998) Mammalian cell microinjection assay to study the function ofRac and Rho. Methods Mol. Biol. 84, 153–160.