Introduction to the EMSA (Gel Shift) Technique
互联网
The interaction of proteins with DNA is central to the control of many cellular processes including DNA replication, recombination and repair, transcription, and viral assembly. One technique that is central to studying gene regulation and determining protein:DNA interactions is the electrophoretic mobility shift assay (EMSA).
The EMSA technique is based on the observation that protein:DNA complexes migrate more slowly than free DNA molecules when subjected to non-denaturing polyacrylamide or agarose gel electrophoresis. Because the rate of DNA migration is shifted or retarded upon protein binding, the assay is also referred to as a gel shift or gel retardation assay. Until conception of the EMSA by Fried and Crothers3 and Garner and Revzin, protein:DNA interactions were studied primarily by nitrocellulose filter-binding assays.
An advantage of studying DNA:protein interactions by an electrophoretic assay is the ability to resolve complexes of different stoichiometry or conformation. Another major advantage for many applications is that the source of the DNA-binding protein may be a crude nuclear or whole cell extract rather than a purified preparation. Gel shift assays can be used qualitatively to identify sequence-specific DNA-binding proteins (such as transcription factors) in crude lysates and, in conjunction with mutagenesis, to identify the important binding sequences within a given gene’s upstream regulatory region. EMSAs can also be utilized quantitatively to measure thermodynamic and kinetic parameters.
The ability to resolve protein:DNA complexes depends largely upon the stability of the complex during the brief time (approximately one minute) it is migrating into the gel. Sequence-specific interactions are transient and are stabilized by the relatively low ionic strength of the electrophoresis buffer used. Upon entry into the gel, protein complexes are quickly resolved from free DNA, in effect freezing the equilibrium between bound and free DNA. In the gel, the complex may be stabilized by “caging” effects of the gel matrix, meaning that if the complex dissociates, its localized concentration remains high, promoting prompt reassociation. Therefore, even labile complexes can often be resolved by this method.
(back to top)
Critical EMSA Reaction Parameters
Target DNA
Typically, linear DNA fragments containing the binding sequence(s) of interest are used in EMSAs. If the target DNA is short (20-50 bp) and well defined, complementary oligonucleotides bearing the specific sequence can be synthesized, purified by gel or HPLC, and annealed to form a duplex. Often, a protein:DNA interaction involves the formation of a multiprotein complex requiring multiple protein binding sequences. In this situation, longer DNA fragments are used to accommodate assembly of multiprotein complexes. If the sequence is larger (100-500 bp), the DNA source is usually a restriction fragment or PCR product obtained from a plasmid containing the cloned target sequence. Protein:DNA complexes formed on linear DNA fragments result in the characteristic retarded mobility in the gel. However, if circular DNA is used (e.g., minicircles of 200-400 bp), the protein:DNA complex may actually migrate faster than the free DNA. Gel shift assays are also good for resolving altered or bent DNA conformations that result from the binding of certain protein factors. Gel shift assays need not be limited to DNA:protein interactions. RNA:protein interactions as well as peptide:protein interactions have also been studied using the same electrophoretic principle.
Labeling and Detection
If large quantities of DNA are used in EMSA reactions, the DNA bands can be visualized by ethidium bromide staining. However, it is usually preferable to use low concentrations of DNA, requiring the DNA to be labeled before performing the experiment. Traditionally, DNA is radiolabeled with 32 P by incorporating an [a -32P]dNTP during a 3´ fill-in reaction using Klenow fragment or by 5´ end labeling using [g -32P]ATP and T4 polynucleotide kinase.
Alternatively, DNA can be labeled with a biotinylated or hapten-labeled dNTP, then probed and detected using an appropriately sensitive fluorescent or chemiluminescent substrate. Pierce offers a chemiluminescent EMSA system (LightShift Chemiluminescent EMSA Kit, Product # 20148 ) and a kit to facilitate labeling DNA with biotin (Biotin 3´ End DNA Labeling Kit, Product # 89818 ). The LightShift EMSA Kit utilizes Pierce’s patented SuperSignal Chemiluminescent Detection Technology to offer detection levels rivaling that of isotopic-based systems.
Nonspecific Competitor
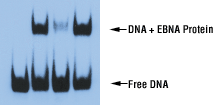
Nonspecific competitor DNA such as poly(dI•dC) or poly(dA•dT) is included in the binding reaction to minimize the binding of nonspecific proteins to the labeled target DNA. These repetitive polymers provide an excess of nonspecific sites to adsorb proteins in crude lysates that will bind to any general DNA sequence. The order of addition of reagents to the binding reaction is important in that, to maximize its effectiveness, the competitor DNA must be added to the reaction along with the extract prior to the labeled DNA target. Besides poly(dI•dC) or other nonspecific competitor DNA, a specific unlabeled competitor sequence can be added to the binding reaction. A 200-fold molar excess of unlabeled target is usually sufficient to out-compete any specific interactions. Thus, any detectable specific shift should be eliminated by the presence of excess unlabeled specific competitor (Figure 16). The addition of a mutant or unrelated sequence containing a low-affinity binding site, like poly(dI•dC), will not compete with the labeled target and the shifted band will be preserved.
| Lane | 1 2 3 4 |
| EBNA Extract | - + + + |
| Unlabeled EBNA DNA | - - + - |
| Unlabeled Oct-1 DNA | - - - + |
 |
Figure 16. EMSA results using the EBNA control system. Biotin-labeled 60 bp duplex bearing the EBNA-1 binding sequence was incubated with an extract in which the EBNA-1 protein was overexpressed. The binding buffer was supplemented with 50 ng/µl poly(dI•dC), 10% glycerol and 0.05% NP-40. Exposure time was 30 seconds with X-ray film.
Binding Reaction Components
Factors that affect the strength and specificity of the protein:DNA interactions under study include the ionic strength and pH of the binding buffer, the presence of nonionic detergents, glycerol or carrier proteins (e.g., BSA), the presence/absence of divalent cations (e.g., Mg2+ or Zn2+), the concentration and type of competitor DNA present, and the temperature and time of the binding reaction. If a particular ion, pH or other molecule is critical to complex formation in the binding reaction, it is often included in the electrophoresis buffer to stabilize the interaction prior to its entrance into the gel matrix.
Gel Electrophoresis
Non-denaturing TBE-polyacrylamide gels or TAE-agarose gels are used to resolve protein:DNA complexes from free DNA. The gel percentage required depends on the size of the target DNA and the size, number and charge of the protein(s) that bind to it. It is important that the protein:DNA complex enters the gel and does not remain in the bottom of the well. Polyacrylamide gels in the range of 4-8% are typically used, although it is not uncommon for higher percentage gels to be used with certain systems. Agarose gels (0.7-1.2%) can be used to resolve very large complexes, as is the case with E. coli RNA polymerase (~460 kDa).
Gels are pre-run at a constant voltage until the current no longer varies with time. The primary reasons for pre-running gels is to remove all traces of ammonium persulfate (used to polymerize polyacrylamide gels), to distribute/equilibrate any special stabilizing factors or ions that were added to the electrophoresis buffer, and to ensure a constant gel temperature. After loading samples onto the gel, it is important to minimize the electrophoretic dead time required for the free DNA to enter the gel matrix, especially when analyzing labile complexes.