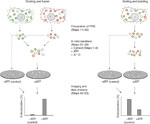
体外荧光法检测核内体早期动力学
互联网
A fluorescence-based in vitro assay for investigating early endosome dynamics
Sina V Barysch1 , 2 , Reinhard Jahn1 & Silvio O Rizzoli2
ABSTRACT
Early endosomes receive material from the plasma membrane by fusion with endocytotic vesicles. This material is sorted within endosomes and directed to subdomains at which carrier vesicles bud. These vesicles are then transported toward different cellular destinations. In this article, we describe a protocol for the cell-free reconstitution of endosome docking/fusion and sorting/budding, which is based on labeling of endosomes by endocytotic uptake with fluorescent cargoes. The protocol includes (i) the preparation of fluorescently labeled endosomes, (ii) assays for docking/fusion and for sorting/budding in vitro and (iii) imaging of the reaction mix by fluorescence microscopy to quantify docking, fusion, cargo sorting and budding using counting of single organelles. Production of endosome stocks requires approximately 1 d. The in vitro reactions can then be performed separately (~1 d) and are conveniently carried out with multiple samples in parallel. The assay can be adapted for studying the dynamics of organelles other than endosomes.
Introduction
Introduction
Early endosomes represent the first sorting station of newly internalized material in all eukaryotic cells. On endocytosis, material is taken up into small carrier vesicles that are targeted to early endosomes, to which they dock and fuse. Endocytosed material is then sorted within the endosome. Depending on the final destination, such cargoes are either packed into budding vesicles, or they remain within early endosomes until they mature into late endosomes and then fuse with lysosomes for degradation of their content. Carrier vesicles budding from early endosomes are delivered to different intracellular destinations such as the plasma membrane, the recycling endosomes or the trans -Golgi network. Thus, early endosomes undergo continuous remodeling due to a steady influx of material by fusion with incoming endocytotic vesicles, which is balanced by an efflux of outgoing vesicles. Moreover, early endosomes frequently fuse with each other (termed homotypic fusion).
Of all these processes, fusion is the only one to be extensively studied. The reason is that fusion can be followed in vitro by biochemical assays relying on contents mixing. In these assays, two sets of endosomes are labeled separately by endocytosis with two markers that possess an intrinsic affinity toward each other (such as antigen and antibody, or avidin and biotin). After cell lysis, endosomes are allowed to fuse in vitro in the presence of an externally added quencher that prevents complex formation between markers escaping from the organelles. The amount of fusion is determined by measuring the degree of binding between markers (reviewed in ref. 1 ). Such assays have defined our current knowledge of fusion dynamics, and most of the proteins involved in homotypic fusion were identified in such in vitro fusion experiments. However, these assays cannot address the vital step that precedes fusion―docking. Furthermore, measuring sorting and budding in vitro has been much more difficult, with most assays relying on the separation of small carrier vesicles from larger donor organelles. For example, permeabilized cells were used to measure endosomal budding of recycling vesicles. In contrast to the endosomal donor organelle, these vesicles can escape from cells though pores in the plasma membrane2, 3 . Moreover, separation through density gradients has been used to study the budding of several types of vesicles from endosome precursors4, 5, 6, 7, 8 . However, these assays only function when there is a substantial difference in buoyancy between the precursor and budded organelles―which explains the paucity of budding assays in the literature.
We have recently developed a new technique for measuring docking and fusion, which is based on the separate labeling of two sets of endosomes with two fluorescent cargo molecules. In vitro fusion of organelles results in mixing of the dyes, producing double-labeled organelles that can be conveniently quantified by fluorescence microscopy9 . Furthermore, we found that measuring the distance between the centers of intensity of the differentially labeled organelles (see Experimental design ) allows for monitoring fusion and docking in parallel10, 11 . Sorting and budding can be investigated by a variation of the technique: organelles are simultaneously labeled with multiple, differently colored markers, and the segregation of markers is followed by monitoring the production of single-color carrier vesicles12 (see Experimental design ).
Our assay has several fundamental advantages over the assays discussed above. First, to the best of our knowledge, our in vitro assay is the only one to date that is able to measure the docking of small organelles quantitatively. Second, it can analyze (and differentiate) docking and fusion in parallel. Third, our budding and fusion assays have no inherent buoyancy requirements, and therefore can be used for investigating virtually any type of organelles (we have already used them for endosomes from various cell types and even synaptic vesicles13 ). Finally, compared with the biochemical in vitro fusion and budding assays, our technique has the advantage of analyzing cargo-containing endosomes on a single-organelle basis. This has allowed, for example, the investigation of protein composition on single sorting organelles, by immunostaining12 . From a technical point of view, the assay can be conveniently upscaled, with up to 20 reactions being run in parallel, in a much easier manner than upscaling, for example, density-gradient assays.
The reliance of the assay on identification of different fluorescently labeled cargoes is an advantage, as indicated above; however, it also introduces several constraints. The fusion of two differently colored organelles is accurately reported, but the fusion of two identically colored organelles is not. In addition, only one double-labeled spot appears when several organelles of different colors fuse together, with the assay thus ignoring multiple fusion events. For the sorting reaction, formation of small vesicles containing both of the fluorescently labeled cargoes is not detected, as they are still double labeled.
Experimental design
Figure 1 . For these assays, cytosol (here we describe the preparation of cytosol from adult rat brains, Steps 1�8) and fluorescently labeled endosomes are needed (we label early endosomes by incubating cultured cells with externally added fluorescent cargo molecules by endocytotic uptake, Steps 9�24). After labeling, cells are cracked with a ball homogenizer and a postnuclear supernatant (PNS), which contains the labeled endosomes, is prepared (Steps 25�30). For docking/fusion assays, two separate sets of differently labeled endosomes are needed. For sorting/budding assays, endosomes are simultaneously labeled with at least two endocytic markers that are sorted into different vesicular pathways. As our laboratory focuses on neuronal membrane trafficking, we use for our assays the neuroendocrine cell line PC12; however, the assay can be adapted to other cell lines.
Figure 1: Experimental design of the in vitro endosome docking/fusion and sorting/budding assay.
For docking/fusion (left), two sets of cells are allowed to take up different fluorescent cargoes (dextran-Alexa 488 and dextran-Alexa 594) by endocytosis for 5 min, leading to the fluorescent labeling of early endosomes with two colors. Cells are cracked and postnuclear supernatants (PNSs) are prepared, which contain fluorescently labeled endosomes. In vitro incubation of the two PNS fractions in the presence of cytosol and an ATP-regenerating system leads to docking and fusion of organelles, which can be visualized by immobilizing them on glass coverslips and subsequent imaging (in which double-labeled organelles appear). Data analysis shows the precise position of each endosome and measures distances between them, thus allowing the quantification of docked and fused endosomes. For sorting/budding (right), cells are simultaneously labeled with two fluorescent cargoes and PNS is prepared. In vitro incubation of the PNS fraction in the presence of cytosol and an ATP-regenerating system leads to cargo sorting and separation because of budding, resulting in a decrease in double-labeled organelles. As for the docking/fusion assay, this can be visualized by fluorescence microscopy and quantified by image analysis.
Full size image (70 KB )In vitro incubation of a reaction mix that contains labeled endosomes, cytosol and an ATP-regenerating system at physiological temperature leads to both docking and fusion, or sorting and budding of the endosomes (Steps 31�39). Depending on the combination of labeled endosomes used, either docking/fusion or sorting/budding is preferentially visualized. The purpose of the assay is to test the function of different molecules in the docking/fusion or sorting/budding processes. Therefore, inhibitors or activators of different molecules should be tested (see Step 37). Various components can be used, from ion chelators (such as calcium- or magnesium-chelating compounds), chemical inhibitors for different processes (such as actin depolymerizing reagents), antibodies for different receptors or effectors (such as clathrin antibodies) to purified effector proteins (e.g., Rab proteins).
To quantify docking/fusion or sorting/budding, we centrifuged the endosomes onto glass coverslips in which they adsorb and can be imaged by epifluorescence microscopy in different color channels (Steps 40�43). Alternatively, it is possible to attach endosomes to coverslips using other approaches such as dilution in low-melt agarose, which is then allowed to harden on the coverslips. For the docking/fusion assay, double-labeled ('yellow') endosomes are visible after incubation. In contrast, sorting/budding leads to the separation of the two labels, resulting in a decrease of double-labeled endosomes.
To distinguish truly double-labeled (fused) from closely apposed (docked) endosomes, we use the following data analysis procedure (Steps 44�53): for each labeled particle a center of intensity is determined in the respective color channel, thus providing information about its precise localization. The distance between the closest neighboring spots (endosomes) in different colors serves as a readout for docking, fusion, sorting and budding. Note that the ability to determine the distance between the two organelles in two channels (i.e., the ability to determine the precise position of each organelle in its respective color channel) is fully independent of diffraction, unlike the situation in which both organelles are similarly labeled. Therefore, this procedure allows information to be obtained beyond the diffraction limit. To correct for drifts between the images acquired in the different channels, we use multicolored fluorescent beads for alignment. The analysis can be performed using various image analysis programs such as Metamorph (Universal Imaging Corporation), ImageJ (NIH), Matlab (The Mathworks Inc.) or Excel (Microsoft Corporation).
As an important control, the procedure should also be performed in conditions in which only multicolor fluorescent beads are added. This allows the determination of the minimal distance between the centers of intensity of perfectly colocalizing objects―a parameter that may vary between different microscopy setups (see Step 50).
Materials
REAGENTS
-
Adult Wistar rats
Caution Experiments involving animals must be carried out in accordance with all appropriate national and institutional regulations. - PC12 cells (clone 251), a similar PC12 line can be obtained commercially from the American Type Culture Collection (ATCC, cat. no. CRL-1721.1)
- Dulbecco's modified Eagles's medium (DMEM; Lonza, cat. no. 12-733F)
- Glutamine (Lonza, cat. no. 17-605E)
- Penicillin-streptomycin (Lonza, cat. no. 17-602E)
- Fetal calf serum (FCS Gold; PAA Laboratories, cat. no. A15-649)
- Horse serum (Biochrom, cat. no. S9135)
- Trypsin/EDTA (Lonza, cat. no. 17-161E)
- TetraSpeck microspheres, 0.2 μm (Invitrogen, cat. no. T7280)
- Fluorescent cargo (all Invitrogen): transferrin-Alexa 488 (T13342), LDL-DiI (L3482), cholera toxin subunit B-Alexa 647 (C34778), dextran-Alexa 488 (D22910) and dextran-Alexa 594 (D22913)
- Protease inhibitors: leupeptin hemisulfate (AppliChem, cat. no. A2183), aprotinin (AppliChem, cat. no. A2132), pepstatin A (Peptide Institute, cat. no. 4039), PMSF (Roth, cat. no. 6367.3), alternatively, Complete EDTA-Free protease inhibitor tablets (Roche, cat. no. 1873580)
- OptiMEM (Invitrogen, cat. no. 31985)
- Creatine phosphate (Roche, cat. no. 621722)
- Creatine kinase (Roche, cat. no. 127566)
- ATP (AppliChem, cat. no. A1348)
- HEPES (Gerbu Biotechnik, cat. no. 1009)
- Magnesium acetate (Merck, cat. no. 1.05819)
- 1,4-DTT (Roth, cat. no. 6908.2)
- Potassium acetate (Merck, cat. no. 1.04936)
- Hexokinase (Roche, cat. no. 1426362)
- Sodium chloride, NaCl (Merck, cat. no. 1.06404)
- Disodium hydrogen phosphate, Na2 HPO4 (Merck, cat. no. 1.06580)
- Sucrose (Merck, cat. no. 1.07687)
- Imidazole (AppliChem, cat. no. A10730)
- Bovine serum albumin, BSA (AppliChem, cat. no. A1391)
- D -Glucose (Merck, cat. no. 1.08342)
- Ethanol (Merck, cat. no. 1.00983)
- Sodium hydroxide, NaOH (Merck, cat. no. 1.06498)
- Potassium hydroxide, KOH (Merck, cat. no. 1.05033)
- Hydrochloric acid, HCl (Merck, cat. no. 1.09057)
EQUIPMENT
- Dissection tools
- Standard cell culture equipment (laminar flow hoods, incubators, cooling centrifuges, water baths, refrigerator and freezers, cell culture plates and flasks)
-
Plastic flasks (15-ml and 50-ml tubes and flasks of different sizes) and plastic pipettes (5, 10, 25 and 50 ml)
Critical All steps must be performed in the absence of detergent traces, i.e., using plasticware or using specially cleaned glassware. -
Centrifuge, Sorvall RC5B Plus, with a SS-34 rotor and polycarbonate flanged tubes (Sorvall, cat. no. 03146)
Critical Tubes must be free of detergent traces. -
Ultracentrifuge, Beckman TL-100, with a TLA-100.3 rotor and polycarbonate tubes (Beckman, cat. no. 349622)
Critical Tubes must be free of detergent traces. - Glass/Teflon Potter Elvehjem Tissue Grinder, 55 ml (OMNI International, cat. no. 07-358054)
- Overhead stirrer (Kika Labortechnik, cat. no. RW20 DZM.n)
- Ball homogenizer (Isobiotech, cat. no. 202 09 547.9, see EQUIPMENT SETUP )
-
Disposable syringe, NORM-JECT (1 ml Ins/TBC; Henke-Sass & Wolf)
Critical This should be free of latex and silicon oils. - Refrigerated table-top centrifuge (Eppendorf, cat. no. 5415 R)
- Reaction tubes, polycarbonate, 0.2 ml (Beckman, cat. no. 343775)
- Lid for reaction tubes: rubber sealing from 1-ml syringes (Dispomed, cat. no. 22009)
- 12-well plates (Corning Costar, 3513)
- 18-mm glass coverslips (Marienfeld Superior, cat. no. 0111580)
- Plate centrifuge (Heraeus Multifuge 4 KR) with a rotor and inlays for plates (Heraeus HIGHplate windshielded rotors)
- Open imaging chamber for 18-mm coverslips (see EQUIPMENT SETUP )
- Epifluorescence microscopy setup (see EQUIPMENT SETUP )
- Bath sonicator (Bandelin Sonorex RK 100)
- Dark (covered) water bath with an agitation device (GFL 1086)
- Forceps
- Pasteur pipettes (plastic)
- Zeiss Axiovert 200M fluorescence microscope (see EQUIPMENT SETUP )
- METAMORPH (Universal Imaging Corporation)
- Matlab (The Mathworks Inc.)
REAGENT SETUP
Critical step All solutions and buffers must be free of detergent and therefore should be prepared in plasticware, or in glassware thoroughly cleaned with ethanol. All reagents that can be stored in aliquots at −20 °C can be used for at least 6 months after preparation.
-
PC12 culture medium
- The medium consists of DMEM supplemented with 10% (vol/vol) horse serum, 5% (vol/vol) fetal calf serum, 4 mM glutamine and 100 U ml−1 each of penicillin and streptomycin. It can be stored at 4 °C and used within 1�2 weeks after preparation.
-
Sucrose buffer
- The solution consists of 320 mM sucrose and 5 mM HEPES, pH 7.4 (adjusted with NaOH). It can be stored at 4 °C and used within 1 week after preparation.
-
Homogenization buffer
- The solution consists of 250 mM sucrose and 3 mM imidazole, pH 7.4 (with HCl). It can be stored at 4 °C and used within 1 week after preparation.
-
PMSF stock solution
- The solution consists of 200 mM PMSF in ethanol. It can be stored at −20 °C and used within 1 week after preparation.
-
Pepstatin A stock solution
- The solution consists of 1 mg ml−1 pepstatin A in DMSO. It can be stored in aliquots at −20 °C.
-
Leupeptin stock solution
- It comprises 10 mg ml−1 leupeptin hemisulfate in H2 O. It can be stored in aliquots at −20 °C.
-
Aprotinin stock solution
- The solution is prepared by diluting 10 mg ml−1 aprotinin in H2 O and can be stored in aliquots at −20 °C.
-
PBS
- The solution contains 150-mM NaCl and 20-mM Na2 HPO4 (disodium hydrogen phosphate), pH 7.4 (with HCl). It can be stored at room temperature (i.e., at 21 °C) for up to 2 months.
-
PBS-BSA
- It should be prepared freshly for PNS preparation and consist of 5 mg ml−1 BSA in PBS.
-
DHM buffer
- The solution consists of 625 mM HEPES, 75 mM magnesium acetate and 10 mM DTT (pH 7.4, with KOH), and can be stored in aliquots at −20 °C.
-
KAc buffer
- The solution consists of 1 M potassium acetate in H2 O and can be stored in aliquots at −20 °C.
-
ATP solution
- The solution consists of 100 mM ATP in H2 O (pH 7.4, with KOH) and can be stored in aliquots at −20 °C.
-
CP solution
- It comprises 800 mM creatine phosphate in H2 O and is stored in aliquots at −20 °C.
-
CK solution
- It comprises 4 mg ml−1 creatine kinase (=3,200 U ml−1 ) in H2 O and is stored in aliquots at −20 °C.
-
Glucose solution
- It consists of 250 mM glucose in H2 O and can be stored in aliquots at −20 °C.
-
Internalization medium
- The internalization medium consists of 1 g D -glucose in one bottle (500 ml) OptiMEM and is stored in aliquots at −20 °C.
EQUIPMENT SETUP
-
Epifluorescence microscopy setup
-
-
The setup consists of a Zeiss Axiovert 200M fluorescence microscope (Carl Zeiss AG) equipped with a ×100, 1.4 numerical aperture oil objective and a CCD camera with a 1,317 × 1,035 Kodak chip (pixel size 6.8 μm × 6.8 μm; Princeton Instruments Inc.). Blue fluorescence is detected using the excitation filter 350/50 D, the beamsplitter 400 DCLP and the emission filter 460/50 D. Alexa 488 fluorescence (absorption/emission maxima ~496/519 nm) is detected with the excitation filter 480/40 HQ, the beamsplitter 505 LP Q and the emission filter 527/30 HQ. DiI fluorescence (absorption/emission maxima ~554/571) is detected using the excitation filter 545/30 HQ, the beamsplitter 570 LP Q and the emission filter 610/75 HQ. Alexa 594 fluorescence (absorption/emission maxima ~590/617 nm) is detected using the excitation filter 560/55 HQ, the beamsplitter 595 LP Q and the emission filter 645/75 HQ. Alexa 647 fluorescence (absorption/emission maxima ~650/665 nm) is detected using the excitation filter 620/60 HQ, the beamsplitter 660 LP Q and the emission filter 700/75 HQ. All filters can be purchased from Chroma. For image acquisition, we use METAMORPH (Universal Imaging Corporation).
-
-
Ball homogenizer
-
14 in great detail. A commercially available variant of the ball homogenizer can be obtained from Isobiotech.
Critical Clearance has to be 20 μm for PC12 cells and may be different for other cells, e.g., 25 μm for BHK cells.
-
14 in great detail. A commercially available variant of the ball homogenizer can be obtained from Isobiotech.
-
Open imaging chamber for 18-mm coverslips
- For imaging, we use a chamber that is manufactured by our in-house instrument shop, consisting of a circular metal base on which a plastic ring is screwed, thus holding the coverslip (see ref. 15 for a detailed description). We use this chamber, as it allows imaging in the presence of buffer. Any other design that allows imaging of coverslips is appropriate.
Procedure
Overview
- Steps 1 - 8 Preparation of rat brain cytosol
- Steps 9 - 10 Culture of PC12 cells
- Steps 11 - 30 Preparation of postnuclear supernatants
- Steps 31 - 39 Preparation of docking/fusion or sorting/budding reactions
- Steps 40 - 43 Imaging of the samples
- Steps 44 - 53 Data analysis
-
Points from here (point 1) up to and including point 8 are related to Preparation of rat brain cytosolTiming : 4�5 h Prepare sucrose buffer with protease inhibitors (1 μg ml−1 pepstatin A (1:1,000 vol/vol of pepstatin A stock solution) and 0.2-mM PMSF (1:1000 vol/vol of PMSF stock solution)) for about 5 ml per rat brain.
Critical step Stir the buffer thoroughly while slowly adding PMSF stock solution, as PMSF might precipitate otherwise. -
Kill 10�40 rats and remove their brains into a beaker with sucrose buffer (without protease inhibitors, kept on ice).
Caution Follow national and institutional guidelines for animal handling. - Wash brains 2�3 times with ice-cold sucrose buffer (without protease inhibitors) to remove most of the blood remaining in the solution.
-
Add 10 brains into the Glass/Teflon Potter Elvehjem Tissue Grinder, add about 5 ml ice-cold sucrose buffer (with protease inhibitors, from Step 1) per brain and homogenize brains by 10 strokes (one stroke consists of one up and down movement) at 900 r.p.m. with an overhead stirrer.
Caution Do not apply too much pressure during the first stroke, as the first homogenization of brains is difficult and should be performed slowly; the glass homogenizer might break otherwise. - Centrifuge homogenate at 3,000g (5,000 r.p.m. in a Sorvall SS-34 rotor) for 10 min at 4 °C. Transfer the supernatant into a fresh tube and discard the pellet.
- Centrifuge the supernatant at 32,000g (16,500 r.p.m. in a Sorvall SS-34 rotor) for 15 min at 4 °C. Transfer the supernatant into a fresh tube and discard the pellet.
- Centrifuge the supernatant at minimum 100,000g (we use 330,000g (90,000 r.p.m. in a Beckman 100.3 rotor)) for 30 min at 4 °C to obtain the cytosol in the supernatant.
- Transfer the cytosol-containing supernatant into a fresh tube and divide into small aliquots (0.3�1 ml), determine the protein concentration using a standard assay16 , snap-freeze the aliquots in liquid nitrogen and store them at −80 °C. The protein concentration of the cytosol concentrations should be in the range between 6 and 10 mg ml−1 .Pause Point If stored at −80 °C, rat brain cytosol can be used for at least 12 months.
-
Points from here (point 9) up to and including point 10 are related to Culture of PC12 cellsTiming : 30 min Split PC12 cells 1:6 into 6�30 plates (∅ 15 cm) using standard procedures for passaging PC12 cells (or any other cell line)17 .
Critical step Dissociate the cells thoroughly, as otherwise clumps may remain that may lead to inefficient cargo uptake. We resuspend the cell pellet in 5 ml fresh PC12 culture medium and break cell aggregates by passing the cells 20 times though a 200-μl pipette tip attached to a 10-ml plastic pipette. -
Allow cells to grow for several days until they reach 80% confluence.
Critical step Cells should reach a high degree of confluence; otherwise, it is difficult to detach them from the plates. - Points from here (point 11) up to and including point 30 are related to Preparation of postnuclear supernatantsTiming : 3�5 h Prepare PBS-BSA (freshly, 30 ml for each labeled PNS) and store on ice, together with thawed aliquots of internalization medium (15 ml per PNS), homogenization buffer (50 ml per PNS) and PBS (10 ml per PNS).
- Prewarm trypsin/EDTA and PBS (37 °C), but store PC12 culture medium in the refrigerator.
- Remove old medium from 6�12 plates of cells and wash each plate with 5 ml PBS.
-
Add 2 ml trypsin/EDTA per plate and incubate at room temperature with strong shaking.
Critical step Cells should be exposed to trypsin/EDTA only until they start detaching from the plates. Longer exposure to trypsin/EDTA might cause the degradation of receptors that are required for the uptake of fluorescent cargoes. -
Stop the reaction with 5 ml cold PC12 medium per plate, collect the cells from the plates (using a 10-ml plastic pipette) and transfer them into 50-ml plastic tubes.
Critical step Cells need to be detached, which may require mechanical force such as repetitively squirting of the medium onto the plate. This procedure is easier when longer trypsin/EDTA incubations have been used (Step 14); it is more efficient in highly confluent cultures (Step 10). - Leave tubes on ice and repeat Steps 13�15 for the next 6�12 plates of cells.
- When all cells are collected, centrifuge all tubes at 250g for 5 min at 4 °C.
- Discard the supernatant and resuspend the cell pellet in 10 ml of ice-cold PBS and transfer to a 15-ml plastic tube. Centrifuge again at 250g for 5 min at 4 °C.
- Discard the supernatant and resuspend the cell pellet in 10 ml ice-cold internalization medium and centrifuge the cells as before (250g , 5 min, 4 °C). If more than one PNS label is needed (e.g., for docking/fusion assay) and two differently labeled PNS fractions are required, resuspend the cell pellet in 20 ml ice-cold internalization medium and divide into equal amounts in two 15-ml plastic tubes before centrifugation (250g , 5 min, 4 °C), for the separate PNSs.
-
Estimate the volume of the resulting cell pellet(s).
Critical step This is just an approximation but is necessary for calculating the amounts of fluorescent cargoes to be added. The volume should be in the range of 0.5�3 ml. - Remove supernatants and warm the pellets at 37 °C for 5 min in a water bath.
-
Add fluorescent cargoes in uptake medium to the cell pellets, as described in the options below, and incubate for 5 min at 37 °C. The medium should have the same volume as the estimated cell pellet(s) and should thus show cargo concentrations twice as high as finally desired (e.g., to label 1 ml cell pellet with 50 μg ml−1 fluorescent transferrin, 1 ml uptake medium with a concentration of 100 μg ml−1 transferrin is required). The cargo composition is dependent on whether the docking/fusion assay (A) or the sorting/budding assay (B) should be used as follows:
-
Docking/fusion assay
- Incubate one set of cells with 500 μg ml−1 dextran Alexa 488, 10 kDa (prepared at 1 mg ml−1 in internalization medium).
- Incubate a second set of cells with 500 μg ml−1 dextran Alexa 594, 10 kDa (prepared at 1 mg ml−1 in internalization medium).
-
Sorting/budding assay
-
For dual labeling with transferrin and LDL: simultaneously incubate cells with 50 μg ml−1 transferrin-Alexa 488 and 3 μg ml−1 LDL-DiI (prepared at 100 μg ml−1 transferrin and 6 μg ml−1 LDL in internalization medium).
Critical step LDL easily oxidizes, which affects the uptake efficiency. The exact concentration may need to be adjusted, with concentrations of up to 10 μg ml−1 being needed after several weeks of storage. To delay the oxidation process, it is recommended to keep the LDL-DiI stock solution (Invitrogen) always sealed with parafilm. - For triple labeling with transferrin, LDL and cholera toxin, incubate cells with 50 μg ml−1 transferrin-Alexa 488, 3 μg ml−1 LDL (both as above) and 3 μg ml−1 cholera toxin subunit B-Alexa 647 (i.e., a concentration of 6 μg ml−1 for the uptake medium). Note that any other combinations of labeled endocytotic cargoes can be used.
-
For dual labeling with transferrin and LDL: simultaneously incubate cells with 50 μg ml−1 transferrin-Alexa 488 and 3 μg ml−1 LDL-DiI (prepared at 100 μg ml−1 transferrin and 6 μg ml−1 LDL in internalization medium).
-
Docking/fusion assay
- Stop the uptake reaction by chilling the cells on ice, centrifuge them at 250g for 5 min at 4 °C and remove the uptake medium.
- Wash the cell pellet(s) three times with 10 ml ice-cold PBS-BSA; centrifuge after each wash at 250g for 5 min at 4 °C.
- Prepare 30�50 ml homogenization buffer with protease inhibitors by adding PMSF, pepstatin A, leupeptin and aprotinin (1:1,000 (vol/vol) of the respective stock solutions).
-
Wash the cell pellet(s) once with ice-cold homogenization buffer and resuspend the pellet(s) in ice-cold homogenization buffer with protease inhibitors (Step 25).
Critical step Use no more than 4× the volume of the cell pellet or the PNSs will be too dilute. -
Crack the cells with a ball homogenizer on ice. Use 1-ml disposable syringes for passing 1 ml of resuspended cells 10�20 times through the precooled ball homogenizer. Repeat the procedure, depending on the volume of cells.
Critical step Before cell cracking, wash the assembled ball homogenizer thoroughly with homogenization buffer (with protease inhibitors, Step 25) to remove air bubbles that would cause damage to the endosomes. -
Transfer the cell homogenate into 1.5-ml tubes and centrifuge at 1,200g for 15 min at 4 °C. This will lead to a separation of the homogenate into two layers: the bottom fraction is the nuclear pellet and the upper fraction is the PNS.
Critical step It is sometimes difficult to see the separation of the two layers properly. Use a bright light source (lamp or bright window) to better illuminate the samples. If no band is visible, further dilute the samples with ice-cold homogenization buffer (with protease inhibitors, Step 25) and repeat the centrifugation process. - Remove the supernatant fractions into a new tube and determine the protein concentration of the PNS using a standard method16 . The PNS concentration is usually between 10 and 16 mg ml−1 .Troubleshooting
- Prepare small aliquots (100�400 μl), snap-freeze in liquid nitrogen and store at −80 °C.Pause Point If stored at −80 °C, PNS fractions can be used for at least 12 months.
-
Points from here (point 31) up to and including point 39 are related to Preparation of docking/fusion or sorting/budding reactionsTiming : 2�4 h Thaw aliquots of PNS (from Step 30), cytosol (from Step 8), DHM buffer, KAc buffer, ATP solution, CP solution, CK solution and homogenization buffer.
Critical step Be careful to thaw PNS and cytosol slowly on ice to avoid unwanted protein degradation or breaking of organelles. This might require up to 30 min, depending on the size of the aliquots.
Critical step Store aliquots of PNS in the dark to avoid bleaching of the fluorescent cargoes. - Sonicate multifluorescent TetraSpeck beads for 2�5 min in the bath sonicator and prepare a 1:1,000 (vol/vol) bead stock solution in PBS (2-μl beads in 2 ml PBS). This can be kept at 4 °C for up to 2 weeks.
- Wash 18-mm glass coverslips in 100% ethanol and place them into 12-well tissue culture plates.
- Dilute bead stock solution (from Step 32) 1:100 (vol/vol) in PBS to obtain the final bead solution and pipette 1 ml of it into each well. Remove the air bubbles that might be present between the plate and the coverslip to avoid coverslips floating on centrifugation.
- Centrifuge plates at 5,880g for 45 min at 4 °C (we use 5,460 r.p.m. in a Heraeus Multifuge); this fixes the beads onto the coverslips. Do not remove the solution after centrifugation.
-
Prepare cytosol mix in the following order on ice, depending on whether an ATP-regenerating system (A) or an ATP-depleting system (B) is required, according to whether a positive control (in presence of ATP) or a negative control (in absence of ATP) is desired:
-
ATP-regenerating system
-
Prepare cytosol mix (volumes correspond to one reaction of 50 μl, scale up accordingly): 100 μg cytosol (usually 10�17 μl, depending on the concentration), 0.9 μl DHM buffer, 2.25 μl KAc buffer, 1.67 μl ATP solution, 1.67 μl CP solution and 1.67 μl CK solution.
The final concentration of the reaction corresponds therefore to 100 μg cytosol, 11.25 mM HEPES, 1.35 mM magnesium acetate, 0.18 mM DTT, 45 mM potassium acetate, 3.3 mM ATP, 26.7 mM creatine phosphate and 6.7 μg creatine kinase (=5.3 U).
-
Prepare cytosol mix (volumes correspond to one reaction of 50 μl, scale up accordingly): 100 μg cytosol (usually 10�17 μl, depending on the concentration), 0.9 μl DHM buffer, 2.25 μl KAc buffer, 1.67 μl ATP solution, 1.67 μl CP solution and 1.67 μl CK solution.
-
ATP-depleting system
- Centrifuge 5 μl hexokinase (1,500 U ml−1 , supplied as ethanol precipitate) at 16,000g for 2 min at 4 °C.
- Remove the supernatant and resuspend the hexokinase pellet in 5 μl glucose solution (250 mM).
-
Cytosol mix (volumes correspond to one reaction of 50 μl; scale up accordingly): 100 μg cytosol (usually 10�17 μl, depending on the concentration), 0.9 μl DHM buffer, 2.25 μl KAc buffer and 5 μl hexokinase-glucose buffer (see above).
The final concentration of the reaction therefore corresponds to 100 μg cytosol, 11.25 mM HEPES, 1.35 mM magnesium acetate, 0.18 mM DTT, 45 mM potassium acetate, 7.5 U hexokinase and 25 mM glucose.
-
ATP-regenerating system
-
Prepare docking/fusion (A) or sorting/budding (B) reactions in 200 μl polycarbonate centrifuge tubes on ice.
Critical step Pipette quickly to avoid warming of the solutions. Certain processes (e.g., docking or aggregation) might take place even during short pipetting times.-
Docking/fusion reaction
- Add cytosol mix (from Step 36, usually between 18 and 25 μl).
- Add chemicals, inhibitory/modulating proteins, peptides, antibodies (optional; see Experimental design for further information).
- Add 100 μg PNS containing dextran-Alexa 488 and 100 μg PNS containing dextran-Alexa 594 (usually between 5 and 8 μl each).
- Adjust to 50 μl with homogenization buffer.
-
Sorting/budding reaction
- Add cytosol mix (from Step 36, usually between 18 and 25 μl).
- Add chemicals, inhibitory/modulating proteins, peptides, antibodies (optional; see Experimental design for further information).
- Add 200 μg PNS containing transferrin-Alexa 488 and LDL-DiI (or other double- or triple-labeled PNS) (usually between 10 and 15 μl).
- Adjust to 50 μl with homogenization buffer.
-
Docking/fusion reaction
-
Close reaction tubes with a lid and incubate reactions for 45 min at 37 °C with a slow agitation (speed: 20 shakes per minute in the GFL 1086 water bath). Leave controls on ice for 45 min.
Critical step Samples should be kept in the dark to avoid bleaching the fluorescent dyes. - Place the samples on ice for some minutes to cool, pipette 7 μl of each reaction onto the bead-containing solution on the coverslips (from Step 35) and centrifuge the 12-well plates at 5,880g (5,460 r.p.m. in a Heraeus Multifuge) for 45 min at 4°C.
- Points from here (point 40) up to and including point 43 are related to Imaging of the samplesTiming : 0.5�6 h Place one coverslip into the open imaging chamber for 18-mm coverslips, cover it with PBS and focus the samples.
-
Acquire images in the channels of the respective label (Alexa 488, DiI, Alexa 594, Alexa 647, depending on the PNS fractions that are used) and in one additional channel in which only fluorescent beads can be visualized (e.g., blue) with an epifluorescence microscope (see EQUIPMENT SETUP ).
Critical step Be careful with bleedthrough between the different channels and adjust the exposure times to minimize it. Troubleshooting </, , body <>