过表达都是相似的,敲除的方法却很多
普健生物(武汉)科技有限公司
我们经常听到这样一句话
“失去了才知道珍惜”。
其实,
只是因为失去了才知道它有多好。
生物学家也利用这种方法来研究基因的功能,
美其名曰
↓
基因敲除(Gene Knockout)是上世纪80年代出现的一种新型遗传工程基因修饰技术,可针对某个特定基因进行改造,令其功能丧失,并研究其对相关生命现象造成的影响,从而推测该基因的生物学功能。
经过近30年的发展,
目前已经出现了很多前沿技术。
当然,
最经典的就数
↓
细胞染色体DNA能够和外源DNA发生重组,用设计的同源片段替代靶基因片段,可达到基因敲除的目的。
主要步骤:
构建基因敲除重组载体
↓
重组DNA导入受体细胞
↓
筛选成功重组的细胞
↓
后续检测
1速度慢,效率低(DNA的同源重组频率一般只有10-6左右):加长两端的同源序列有助于重组,但加大了构建基因敲除载体的难度。该技术主要通过大量培养来增加获得敲除转化子的概率。
2载体转化和表达比较关键,周期较长,对细胞感受态要求也较高。
3对于真核生物的同源重组基因敲除研究较少:真核生物转化不容易,表达周期长,基因调控复杂,敲除之后不一定出现特定表型。
另外,
也有一些
Red同源重组系统
Red同源重组系统主要包含3种蛋白质分子,分别称为Exo、Beta和Gam,这三者是λ噬菌体中的高效重组蛋白质。其中Exo蛋白是一种核酸外切酶,结合在双链DNA的末端,从5'端向3'端降解DNA,产生3'突出端;Beta蛋白结合在单链DNA上,介导互补单链DNA退火,Gam蛋白可与RecBCD酶结合,抑制其降解外源DNA的活性。
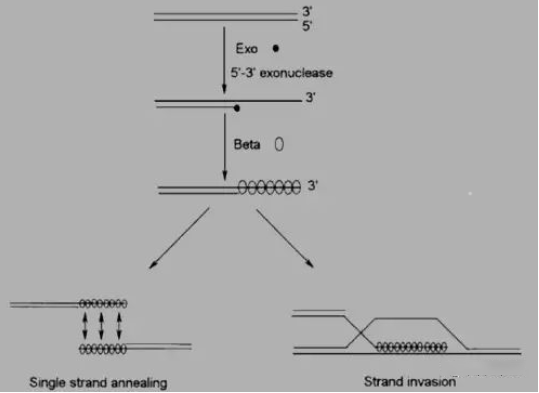
▲
Red 同源重组流程
此外,还有RecA重组敲除系统(由RecA和RecBCD蛋白组成,在基因敲除时必须以环状质粒存在)、结合转化敲除系统及温度敏感型质粒敲除系统等可供选择。
FLP-FRT系统和Cre-loxP系统
这两个系统都是位点特异性重组酶系统,其中FLP-FRT源于酿酒酵母,Cre-loxP源于F1噬菌体。两个系统的基本原理类似。以Cre-loxP系统为例,该系统含有两种成分:第一个部分是一段长34 bp的重组酶识别的DNA序列(loxP),其中含有两个13 bp的反向重复序列和一个8 bp的核心序列;第二个是Cre重组酶,可以引发loxP位点的DNA重组。当靶基因位于两个loxP位点之间时,在Cre重组酶的作用下,如果两个loxP位点的方向相同,则靶基因被敲除;如果两个loxP位点的方向相反,则靶基因的方向发生倒转。

▲
Cre-loxP系统基因敲除原理
基因捕获技术
基因捕获(Gene Trap)是功能基因组学的研究方法,应用于表达基因的寻找。
其原理主要是:
利用随机插入突变进行基因敲除。基因捕获载体中包括一个无启动子的报道基因,只有转化到有启动子的(能够表达的)DNA序列上,才能够表达报告基因。结合cDNA文库和DNA芯片,由此便可以报告一个功能基因的位置。因此,基因捕获也可以当做基因敲除文库来使用。现在已经得到了基因捕获体系的大肠杆菌文库,可以进行很多细菌分子生物学实验和分析。
基因捕获的优缺点是并存的。
优点:用常规方法进行基因敲除研究需耗费大量的时间和人力,研究者必须针对靶位点在染色体组文库中筛选相关的染色体组克隆,绘制相应的物理图谱,构建特异性基因敲除载体及筛选靶细胞等,而利用基因捕获能够节省大量筛选染色体组文库以及构建特异打靶载体的工作及费用,能更有效和迅速地进行染色体组的功能分析。
缺点:一是它只能够敲除表达的基因,难以对深入的调控问题做研究。二是无法对基因进行精细的遗传修饰。
再来看一下
↓
反义RNA和RNAi技术
反义RNA是指与mRNA互补的RNA分子,也包括与其他RNA互补的RNA分子。由于核糖体不能翻译双链的RNA,所以反义RNA与mRNA特异性地互补结合, 即抑制了该mRNA的翻译。
根据反义RNA的作用机制可将其分为3类:
Ⅰ类反义RNA直接作用于靶mRNA的SD序列和部分编码区,直接抑制翻译,或与靶mRNA结合形成双链RNA,从而易被RNA酶 Ⅲ 降解;
Ⅱ类反义RNA与mRNA的非编码区结合,引起mRNA构象变化,抑制翻译;
Ⅲ类反义RNA则直接抑制靶mRNA的转录。
双链RNA进入细胞后,能够在Dicer酶的作用下被裂解成siRNA,而另一方面双链RNA还能在特定聚合酶下形成单链,并和某些蛋白形成复合物使mRNA被RNA酶裂解,并且以siRNA作为引物,以mRNA为模板形成双链RNA。最后形成siRNA聚合酶链式反应,显著增加对基因表达的抑制。
RNAi敲除主要流程
调研目的基因的mRNA序列
↓
设计DNA片段编码反义RNA
↓
利用基因重组构建人工表达载体
↓
载体转化至宿主细胞,表达反义RNA
↓
筛选表型和后续基因功能研究
RNAi基因敲除没有对原来的基因做出改动,因而目的基因在细胞内是完好存在的,并且RNAi现象普遍,在几乎所有真核生物中都能发挥作用。该技术已被广泛应用于基因功能探索和传染性疾病及恶性肿瘤的基因治疗。
-
比用同源重组法更加简便,缩短周期。
-
对于一些敲除后外培养的细胞,利用RNAi技术研究它们的功能。
-
由于RNAi能高效特异地阻断基因的表达,因此是研究信号传导通路的良好工具。
-
容易调控,可用来研究生长发育过程中起作用的基因。
-
序列必须已知,反义DNA必须人工合成,并且序列的选择原则尚不清楚。
-
目前siRNA导入胞内的效率低下,如何有效将siRNA转移入体内成为RNAi应用的最大障碍。
-
基因沉默的时效性和遗漏,尤其在多基因家族中的非特异性问题尚未得到解决。
-
目前RNAi 机制尚未完全阐明,因此需要更加深入的研究。
还有
ZFNs(锌指核糖核酸酶)是由一个DNA识别域和一个非特异性核酸内切酶构成。DNA 识别域是由3-6个Cys2-His2锌指蛋白(zinc-fingers)串联组成,每个锌指蛋白识别并结合一个特异的三联体碱基,形成α-β-β二级结构。其中α螺旋的16氨基酸残基决定锌指的DNA结合特异性,骨架结构保守。对决定DNA结合特异性的氨基酸引入序列的改变可以获得新的DNA结合特异性。根据目的基因设计8-10个锌指结构域并与FokⅠ核酸内切酶结合可构成靶向特定位点的ZFNs,在FokⅠ切割域的二聚化作用下完成对目的基因的编辑。
TALEs蛋白包括三部分:N端序列、C端序列、由高度保守的重复单元组成的中心重复区。每个重复单元中除12、13位氨基酸(repeat varible di-residues,RVDs)可变外其余几乎相同,RVD与A、T、C、G之间有着严格的对应关系,即NI-A;NG-T;HD-C;NN-G。构建TALE时,根据靶DNA序列变换RVDs并串联重复单元并与FokI切割结构域结合。TALE与靶位点结合,二聚化的FokI对其进行切割,完成基因编辑操作。
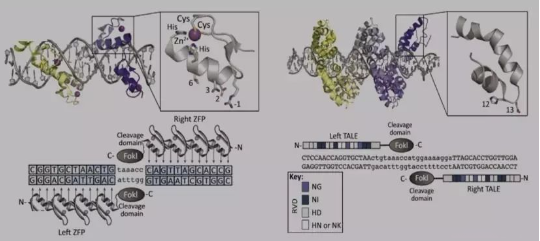
▲
优点和局限
ZFN 能够识别并结合指定的基因序列位点,并高效精确地切断。随后细胞利用天然的 DNA 修复过程来实现 DNA 的插入、删除和修改。这项技术中设计特异性的ZFN是最关键的环节,可结合计算生物学方法设计高特异性的ZFN,但面临的挑战是难以完全找到匹配的3联子锌指,并且脱靶效应(Off-target)严重。
TALE的DNA结合域中重复氨基酸序列模块可以与单碱基发生特异性结合,TALEN 技术能够克服ZFN方法不能识别任意目标基因序列以及识别序列经常受上下游序列影响等问题,因此灵活性更好,并且基因操作简单方便,但脱靶效应同样严重。
更有兴起不久的
↓
CRISPR/Cas9系统
CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats)/Cas( CRISPR-associated) 系统是一种广泛存在于细菌与古细菌中,由RNA介导的可遗传获得性免疫系统。它能够为宿主细胞提供对外源DNA(如噬菌体质粒)的免疫功能。在此基础上建立起来的CRISPR/Cas9 基因敲除系统目前广泛应用于原核生物、酿酒酵母、线虫、斑马鱼及哺乳动物中。
工作原理
crRNA(CRISPR-derived RNA)通过碱基配对与tracrRNA(trans-activating RNA)结合形成 tracrRNA/crRNA 复合物,引导核酸酶 Cas9蛋白在与crRNA 配对的序列靶位点剪切双链DNA。细胞通过自身的同源重组或者非同源重组机制修复受损的双链DNA,从而达到基因敲除的目的。研究者通过改造形成更加简洁的具有引导作用的sgRNA(short guide RNA),也能够引导Cas9对DNA的定点切割。
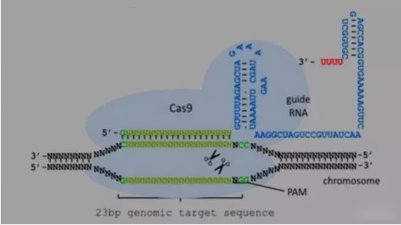
▲
Cas/gRNA复合体结构及基因剪切机制
主要流程
寻找靶基因中合适的gRNA序列
↓
设计单链oligo序列,退火形成双链DNA
↓
利用基因重组将双链连到表达载体
↓
载体转化至宿主细胞
↓
筛选阳性克隆
↓
gRNA引导Cas9蛋白对靶位点进行剪切
↓
克隆子测序验证
↓
筛选表型和后续基因功能研究
sgRNA设计网址大奉送
http://spot.colorado.edu/slin/cas9.html
http://www.ecrisp.org/E‐CRISP/;
http://zifit.partners.org/ZiFiT/ChoiceMenu.aspx
http://crispr.mit.edu/
http://tools.genome-engineering.org
http://cas9.cbi.pku.edu.cn/
http://cbi.hzau.edu.cn/cgi-bin/CRISPR
http://crispr-era.stanford.edu/
https://crispr.bme.gatech.edu/
写在最后
基因敲除技术在生命科学及相关领域中应用非常广泛,并且在不断发展和完善中,最终目的是让靶基因的表达降为零。这可以通过基因表达调控的多个层面进行,如基因的转录、终止、转录后调控、翻译、翻译后调控、酶活性调控等。
现阶段的基因敲除技术主要建立在基因的转录和翻译等层次并始终面临着巨大困难,其原因有二:
-
基因有着复杂的表达和调控机制,生物体中很多基因行使着相同的功能,敲除一个功能基因后并不一定能够引起明显的改变和出现容易识别的表型(曾报道过将酵母菌40%的基因沉默掉,它仍能够正常生长)。
-
另一方面,对于某些必需基因或看家基因(House-keeping Gene),敲除后会影响细胞的生长或造成细胞的死亡,导致无法对这些基因进行相应的研究。
因此,
基因敲除技术
始终伴随着其他相关技术前进着,
在生命知识揭示和发现过程中
起着举足轻重的作用。