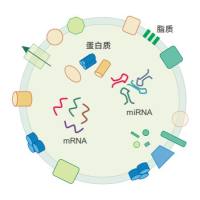
分子生物学产品常见问题分析
互联网
1325
|
分子生物学产品常见问题分析
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
三、DNA分子量标准问题分析:
以λ/Hind III为代表的λDNA酶切片段用作电泳分析中的分子量标准,会发现电泳后分子量条带不对的问题,解释原因如下:在Lambda DNA分子的左右臂存在着12个碱基组成的具有互补顺序的单链突出端-COS位点,COS位点的退火复性,在λDNA片段电泳分离时会出现问题,含左右臂片段在胶上应出现的地方条带会减弱或完全看不到,而复性的左右臂片段会结合成大的片段,即在较大的分子量区域内产生一个额外的条带,这就造成了不正常的带型。解决以上问题的办法,是在电泳之前,把DNA分子量标准67℃加热约10分钟,然后立即放入冰浴中骤冷,待样品冷却后上样。这样即可解离复性的粘性末端,使电泳过程DNA片段形成正常的带型分布。
四、凝胶电泳操作注意事项: 1. 缓冲系统:在没有离子存在时,电导率最小,DNA不迁移,或迁移极慢,在高离子强度的缓冲液中,电导很高并产热,可能导致DNA变性,因此应注意缓冲液的使用是否正确。长时间高压电泳时,常更新缓冲液或在两槽间进行缓冲液的循环是可取的。 2. 琼脂糖:不同厂家、不同批号的琼脂糖,其杂质含量不同,影响DNA的迁移及荧光背景的强度,应有选择地使用。 3. 凝胶的制备:凝胶中所加缓冲液应与电泳槽中的相一致,溶解的凝胶应及时倒入板中,避免倒入前凝固结块。倒入板中的凝胶应避免出现气泡,影响电泳结果。 4. 样品加入量:一般情况下,0.5cm宽的梳子可加0.5ug的DNA量,加样量的多少依据加样孔的大小及DNA中片段的数量和大小而定,过多的量会造成加样孔超载,从而导致拖尾和弥散,对于较大的DNA此现象更明显。 5. 电泳系统的变化会影响DNA的迁移,加入DNA标准参照物进行判定是必要的。 6. DNA样品中盐浓度会影响DNA的迁移率,平行对照样品应使用同样的缓冲条件以消除这种影响。 7. DNA迁移率取决于琼脂糖凝胶的浓度,迁移分子的形状及大小。采用不同浓度的凝胶有可能分辨范围广泛的DNA分子,制备琼脂糖凝胶可根据DNA分子的范围来决定凝胶的浓度。小片段的DNA电泳应采用聚丙烯酰胺凝胶电泳以提高分辨率。 |