差异表达基因克隆技术的新进展
互联网
- 相关专题
崔大祥 闫小君 苏成芝 2004-8-2 16:34:54 国外医学遗传学分册1999年第22卷第5期
提要 分离并克隆 差异表达基因是目前功能性基因研究的热点。mRNA差异展示是筛选差异表达基因最有效的技术之一,它的主要不足是有一定假阳性。RDA、SSH、DSD、LSH技术能够克服假阳性,但也具有一定的不足,应根据需要选择运用。随着能扩增长片段及具有自动校正功能的Taq酶出现及应用,这些技术将会不断完善与发展,具有广阔的应用前景。
关键词 差异表达基因;克隆
现代分子生物学研究表明,人类基因组 约由10万左右的不同基因组成,这些基因选择性的表达决定了机体整个生命过程,基因表达的变化处于控制生物学调节机制的中心位置。因此,分离并克隆差异表达基因不仅有助于阐明生命的粤秘,而且还能为基因诊断与治疗提供重要的理论依据。近几年来,差异表达基因克隆技术不断完善与发展,已成为研究肿瘤和疾病等相关基因的重要手段。
一、mRNA差异展示
mRNA差异展示(mRNA differential display,DDRT-PCR )是目前筛选差异表达基因最有效的方法之一,其基本原理是:3´端采用锚定引物,与mRNA3´poly a结合,进行反转录,形成mRNA:cDNA杂交分子,5´端采用20条随机引物,与锚定引物一起进行PCR扩增,其产物经测序胶显示出来,再回收鉴定克隆差异条带。1992年Liang和Pardee[1]建立此技术时,5´端采用20条随机引物,每条由10个碱基组成,3´端采用12条引物即T12MN(M=A、C或G,N=A、G、C、T)。这种组合从统计学上几乎能完全覆盖所有的mRNA序列,差异表达的基因会全部呈现出来。实践证明,这种方法有效,但假阳性率在50%~70%左右,差异条带在Northern印迹上重现性差,后续处理也比较复杂。1994年Ito[2]等对3´端引物进行了改进,把12条引物改为3条,即T12A、T12C、T12G,使得实验操作简化。1995年Ayala[3]等提出了在3´端锚定引物与5´端随机引物上皆增加10个碱基,使得上下游引物Tm值在60℃左右,PCR循环参数也改为前5个循环采用Liang和Pardee推荐的参数,即94℃30s,40℃2min,72℃30s。经这样改进,显著地增强了差异条带的重复性和敏感性,同时使得差异条逼带的克隆十分方便。1996年4月,Liang和Pardee[4]对5´端此物进行了改进,引物条数改为8条,皆带上HindⅢ酶切位点,每条由13个碱基组成,3´端锚定引物采用3条,也带上HindⅢ酶切位点,每条由18个碱基组成,PCR循环条件仍采用94℃30s,40℃2min,72℃30s,40个循环,最后在72℃延伸7min。1998年[5]我们在Liang和Pardee改进的基础上,对PCR循环参数进行了改进,即前5个循环采用94℃30s,40℃2min,72℃30s,后35个循环采用94℃30s,55℃2min,72℃30s,最后在72℃延伸7min。经这样改进,既保持了扩增效率,又增强了差异条带的特异性及重复性,降低了假阳性率。
总之,mRNA差异展示具有简便、快速、所需起始材料少、同时可在多个材料之间或不同处理材料之间进行比较等优点,但具有较高频率的假阳性和短小的差异片段的缺点,给后续处理带来一系列问题,虽然此技术经过了一些起改进,但这两个缺点仍限制着此方法的充分应用。
二、代表性差示分析
代表性差示分析(representational difference analysis,RDA)最早由Lisitsyn等提出的。1994年,Hubank和Schatz[6]将其应用于克隆差异表达基因,其基本原理是:靶方(Tester,T)和驱动方(Driver,D)的cDNA在进行差方式分析前均用一种识别4碱基序列的限制酶作切割处理,形成平均长度为256bp的cDNA片段代表群,第一次T与D杂交反应时,两者总量比为1:100,第二次增加到1:400,第三次增加到1:8000,再利用PCR以指数形式扩增双链模板,而仅以线性形式扩增单链模板这一特性,通过降低cDNA群体复杂度和多次更换cDNA两端接头,来特异扩增目的基因片段。特别是T减D杂交反应后仅设置了72℃复性与延伸及95℃变性这两个循环参数,共20个循环,从而使PCR产物特异性大大提高。此技术最大的优势是差异条带在Northern印迹上重现性好,假阳性少。缺点是所需起始材料较多,更多信赖于PCR技术,T与D之间若存在较多差异,或T中存在某些基因上调表达,则难达到预期目的,而且工作量比DDRT-PCR 大,周期长。
三、抑制性扣除杂交
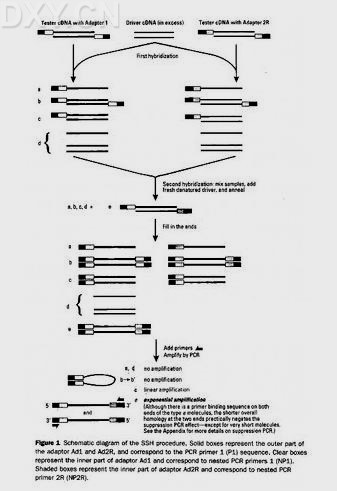
抑制性扣除杂交(suppression subtractive hybridization,SSH),是1996年Diatchenk[7]等提出的一种新的克隆基因技术,其基本原理是:先将T与D方cDNA用限制酶切割为小片段,将T方平均分为两份,分别连接不同的接头,然后与过量的D方cDNA进行不充分杂交。根据复性动力学原理,浓度高的单链分子迅速复发,而浓度低的单链分子仍以单链形式存在。然后混合两份杂交样品,同时加入新的变性D方cDNA进行第二次扣除杂交,杂交完全后补平末端,加入合适引物接头(接头1与接头2引物)进行PCR扩增。当DNA两条链含相同接头时,其PCR扩增受到抑制,而含不同接头的双链DNA分子才可进行指数扩增,其产物即为目的片段。
此技术避免了消减杂交过程中分离单双链DNA的步骤,可成千倍地扩增目的片段,能分离出T方上调表达的基因,与DDRT-PCR相比,具有假阳性少重复性强的优点,它的最大不足就是需要较多的起始材料,更多依赖于PCR技术,不能同时进行数个材料之间或不同处理材料之间的比较。