基因差异表达技术
互联网
真核生物中,从个体的生长、发育、衰老、死亡,到组织的得化、调亡以及细胞对各种生物、理化因子的应答,本质上都涉及基因的选择性表达。
高等生物大约有30000个不同的基因,但在生物体内任意8细胞中只有10%的基因的以表达,而这些基因的表达按特定的时间和空间顺序有序地进行着,这种表达的方式即为基因的差异表达。其包括新出现的基因的表达与表达量有差异的基因的表达。
生物体表现出的各种特性,主要是由于基因的差异表达引起的。
由于基因的差异表达的变化是调控细胞生命活动过程的核心机制,通过比较同一类细胞在不同生理条件下或在不同生长发育阶段的基因表达差异,可为分析生命活动过程提供重要信息。
研究基因差异表达的主要技术有差别杂交(differential hybridization)、扣除(消减)杂交(subtractive hybridization of cDNA ,SHD)、mRNA 差异显示(mRNA differential display,DD)、抑制消减杂交法(suppression subtractive hybridization,SSH)、代表性差异分析(represential display analysis,RDA)、交互扣除RNA 差别显示技术(reciprocal subtraction differential RNA display)、基因表达系列分析(serial analysis of gene expression,SAGE)、电子消减(electronic subtraction)和DNA 微列阵分析(DNA microarray)等。
一、差别杂交与扣除杂交
差别杂交(differential hybridization)又叫差别筛选(differential screening),适用于分离经特殊处理而被诱发表达的mRNA 的cDNA 克隆 。为了增加这种方法的有效性,后来又发展出了扣除杂交(subtractive hybridization)或扣除cDNA (subtractive cDNA cloning),它是通过构建扣除文库(subtractive library)得以实现的。
(一)差别杂交
从本质上讲,差别杂交也是属于核酸杂交的范畴。它特别适用于分离在特定组织中表达的基因、在细胞周期特定阶段表达的基因、受生长因子调节的基因、以及在特定发育阶段表达的或是参与发育调节的基因,同时亦可有效地用来分离经特殊处理而被诱发表达的基因。目前,差别杂交筛选法在基因的分离工作中有着相当广泛的用途。
差别杂交的技术基础十分简单,它不需要任何有关的目的基因的核苷酸序列信息,而重要的是耍拥有两种不同的细胞群体:在一个细胞群体中目的基因正常表达,在另一个细胞群体中目的基因不表达。在这种情况下便可制备到两种不同的mRNA 提取物。
其一是含有一定比例的目的基因mRNA 类型的总mRNA 群体,其二是不含有目的基因mRNA 类型的总mRNA 群体。因此,可以通过这两种总mRNA (或是它们的cDNA 拷贝)为探针的平行杂交,对由表达目的基因的细胞总mRNA 构建的库进行筛选。
当使用存在目的基因的mRNA 探针时,所有包含着重组体的菌落都呈阳性反应,在X光底片上呈现黑色斑点,而使用不存在目的基因的mRNA 探针时,除了含有目的基因的菌落外,其余的所有菌落都呈阳性反应,在X光底片上呈现黑色斑点。比较这两种底片并对照原平板,便可以挑选出含目的基因的菌落,供作进一步研究使用。
差别杂交筛选技术已被成功地用于分析爪蟾和粘菌的发育问题。这两个应用例子表明,处于不同发育状态或阶段的丰度相差5倍的特异的mRNA 种是能够被检测出来的。生长因子调节基因(growth factor-regulated gene)的克隆 ,是差别杂交成功应用的一个典型例子。
我们知道,血清中含有生长因子,因此用血清处理处于静止期的细胞时,便会迅速诱发生长因子调节基因进行表达。所以,分别从静止期细胞培养物和经血清激活3小时的物中提取的poly(A)mRNA 制剂,在mRNA 种类上是有差别的,至少后者比前者多出了一种生长因子调节基因的mRNA 类型。
用从激活细胞中分离的poly(A)mRNA 反转录合成的cDNA 与λ噬菌体载体重组,构成cDNA 文库,并同时复制两份硝酸纤维素滤膜。A组滤膜同血清激活细胞制备的cDNA 探针杂交,B组滤膜同静止期细胞制备的cDNA 探针杂交。
将所得的放射自显影图片进行仔细的比较,从中鉴定出只同激活细胞探针杂交而不能同静止期细胞探针杂交的噬菌斑位置。这些便有可能是带有受血清诱发表达的生长因子调节基因的DNA 编码序列。
(二)扣除杂交
差别杂交可有效地对于因特殊处理而被诱发产生的mRNA 的cDNA 克隆 的分离,或是在细胞中具高表达效率的mRNA 之cDNA 克隆 的分离,但对于低丰度的mRNA 的cDNA 的分离则有相当的困难。为了进一步提高差别杂交的筛选效率,一种切实可行的办法是应用扣除杂交筛选法构建富含目的基因序列的cDNA 文库。
扣除杂交法的本质是除去那些普遍共同存在的、或是非诱发产生的cDNA 序列,从而使待分离的目的基因的序列得到有效的富集,提高了分离的敏感性。下面以T细胞受体(T-cell receptor,TCR有时亦称之为T细胞抗原受体)编码基因的分离为例子,说明扣除杂交筛选法的基本原理与简要过程。
T细胞和B细胞来自共同的前体细胞,两者都能够识别特异的抗原。但与B细胞不同,T细胞不能识别游离的抗原,而只能识别在其它细胞表面的抗原。T细胞的这种抗原识别特异性是由TCR基因决定的。TCR基因只能在T细胞中表达,而不能在B细胞中表达。
那么从T细胞mRNA 制备来的单链cDNA ,同大大超量的B细胞的mRNA 在有利于发生DNA -RNA 杂交的条件下保温。
其结果会是所有的能够在T和B两类细胞中同时表达的T细胞基因的cDNA 分子(约占98%),都能与B细胞的mRNA 退火形成DNA -RNA 杂交分子,而不能在B细胞中表达的、T细胞特有的cDNA (约占2%),由于B细胞中没有相应的mRNA ,故不能形成DNA -RNA 杂交分子,仍然处于单链的状态。
将此种杂交混合物通过羟基磷灰石柱(hydroxylapatite column),于是DNA -RNA 杂交分子便结合在柱上,而游离的单链cDNA 则过柱流出。回收到的T细胞特异的cDNA 被转变为双链cDNA 之后,与适当的λ噬菌体载体重组并转染给大肠杆菌寄主细胞,这样便得到了T细胞特异cDNA 高度富集的扣除文库。
然后再按照同样方法制备扣除的cDNA 探针,即被B细胞mRNA 杂交扣除了的T细胞特异的cDNA 探针,筛选文库,可成功地分离到了T细的TCR基因。
扣除杂交法同样也可以用来分离缺失突变基因。从野生型植株制备的染色体总DNA ,用一种适当的核酸内切限制酶(比如Sau 3A)切割成小片段。同时从缺失突变体植株制备的染色体总DNA ,经随机切割之后,用生物素(biotin)进行标记,作为非同位素标记探针使用。
取大大超量的此种探针,同Sau 3A酶切的野生型染色体总DNA 片段混合,经变性、退火处理,溶液中的无生物素标记的野生型的DNA 分子便同生物素标记的突变型的DNA 探针杂交。将杂交反应混合物通过生物素结合蛋白质柱(avidin column)。
这种柱是用包裹着生物素结合蛋白质的专用的细小磁珠装填的。大部分野生型植株的DNA 分子都同突变型植株的生物素标记的DNA 探针杂交,便被结合到柱上。而野生型植株的DNA 片段由于在突变型DNA 中缺失了相应的片段,故没有相应的生物素标记的探针与之杂交,经洗脱便过柱流出。
随后将洗脱收集的DNA 同超量的生物素标记探针再杂交,再过柱。如此经过多次重复富集之后,用PCR法扩增DNA 片段,并予以克隆 。最后用Southern杂交法进一步鉴定出,只同野生型DNA 杂交而不能同突变型DNA 杂交的含有突变基因的阳性。
(三)差别杂交的局限性和扣除杂交的缺点
实践表明,应用差别杂交技术分离克隆 的目的基因存在着诸多方面的局限性。首先,差别杂交的灵敏度比较低,特别是对于那些低丰度的mRNA 而言,这个缺点就显得更加突出。这是因为在差别杂交中所使用的杂交探针是mRNA 反转录成的cDNA 群体。在这些同位素标记的探针中,真正能与目的基因核苷酸序列完全互补的仅占很低的比例,以至于那些低丰度的mRNA 之cDNA 克隆 ,是很难用此法检测出来的。其次,差别杂交需要筛选大量的杂交滤膜,鉴定大量的噬菌斑或克隆 片段,因此是十分耗费时间和金钱的工作。况且两套平行转移的滤膜之间,DNA 的保有量往往是有差别的,这样所得的杂交信号的强度也就不会一致,需要重新进行点杂交,以作进一步的阳性鉴定工作。所以说重复性差是差别杂交筛选法的又一个缺点。
扣除杂交技术在理论上很具吸引力,但实际操作并不容易。首先是回收cDNA 量有限,并仍存在重复性差、敏感度低的缺点,这些均限制了这一技术更广泛的使用。
二、mRNA 差别显示技术(DDRT-PCR )
随着PCR技术的发展,人们在此基础上建立起了一系列基于基因分离的新技术新方法。如mRNA 差别显示技术(DDRT-PCR )、以及进一步改进的代表性差示分析(RAD),抑制性扣除杂交(SSH)和交互扣除RNA 差别显示技术(RSDD)。
差别显示PCR是根据绝大多数真核细胞mRNA 的3′端具有的多聚腺苷酸尾(polyA)结构,因此可用含oligo(dT)的寡聚核苷酸为引物将不同的mRNA 反转录成cDNA 。该方法的创始人Liang P和Pardee A根据Poly A序列起点前2个碱基除AA外只有12种可能性的特征,设计合成了12种下游引物,称3′-锚定引物,其通式为5′-T11MN;同时为扩增出polyA上游500 bp以内所有可能性的mRNA 序列,在5′端又设计了20种10 bp长的随机引物。这样构成的引物对进行PCR扩增能产生出20000条左右的DNA 条带,其中每一条都代表一种特定mRNA 类型,这一数字大体涵盖了在一定发育阶段某种细胞类型中所表达的全部mRNA 。回收不同组织所特有的差别表达条带中的DNA ,再扩增至所需含量,进行Southern blot或Northern blot或直接测序 ,从而对差异条带鉴定分析,以便最终获得差异表达的目的基因。
差别显示技术自1992年建立后,一直在不断进行着改进。如在1994年,Ito等对3′端锚定引物的设计由固定两个碱基变为一个碱基固定的引物,这就使原来12种引物减至3种即可(5′T12G,5′T12A,5′T12C),这样做减少了每个mRNA 样品对逆转录反应种类的需要,并且把由于简并性引起的某些RNA 的代表性差和RNA 数量过多现象降低到最低程度;在随后的两年中,研究人员有在3′端引物和5′随机引物末端分别加上了限制性内切酶识别位点(如Hind III酶切位点),使得5′端引物条数改为8条,长度为13 bp,而3′端引物则由18个碱基组成。这样形成的24种引物对,经计算机同源性分析表明同样能覆盖全部mRNA ,使实验简化,同时由于引物变长,使cDNA 扩增更为有效。有的实验室采用把Bakman公司的kit,其引物5′端为26 bp,3′端为31 bp,并加上了T3和T7两端的测序 引物,由仪器切割差异条带后,再次扩增,并通过荧光标记,用计算机统计出结果。此外,许多学者针对该技术在操作中假阳性高等问题,还从设计对照、提取胞质RNA 、更换放射性标记物以及改变PCR反应条件等等方面提出了相应解决方案,以求这一技术得到进一步完善。
mRNA 差别显示技术(DDRT-PCR )与示差筛选、扣除杂交相比,具有很多优点:①速度快,较易操作;②由于PCR扩增技术的应用,使得低丰度mRNA 的鉴定成为可能,且灵敏度高,③可同时比较两种以上不同来源的mRNA 样品间基因表达的差异。
尽管差别显示技术有以上诸多方面的优点,但在实际操作中仍存在一些问题,主要表现在:①出现差别条带太多,假阳性率高达70%左右,重复性差,且对高拷贝数的mRNA 具很强的倾向性;②扩增条带分子长度较短,一般在110-450 bp之间。
三、代表性差异(RAD)和抑制性扣除杂交(SSH)
(一)代表性差异(RAD)
代表性差示分析,充分发挥了PCR以指数形式扩增双链模板,而以线性形式扩增单链模板的特性,通过消减和富集,使得目的基因片段得到特异性扩增。将待测cDNA (Tester)和驱动cDNA (Driver)分别用同一种经识别4碱基序列的限制性内切酶切割形成的平均长度为256 bp的cDNA 片段代表群,保证了绝大多数遗传信息均能被PCR扩增;分别接上寡聚核苷酸接头(adaptor),并以接头为引物进行PCR扩增,此步将大小不等的DNA 片段分离纯化;将接头切除,并只在Tester片断末端接上新接头,然后将Tester与大大过量的Driver混合杂交。Driver过量的目的是使T群体中特异性序列没有遗漏的可能;复性,补平末端,并以新接头为引物进行PCR扩增,形成的3种杂交体中只有自身退火形成的Tester/Tester两端均能和引物配对,产物双链DNA 数量呈指数递增。Tester/Driver杂交体只能是单引物扩增,产物为单链DNA 分子,其数量呈线性递增。Driver/ Driver杂和体由于分子两端没有与新引物配对区而无法进行扩增;用Mung Bean Nuclease去除单链DNA 分子,差异双链cDNA 便完成第一轮富集。实验中使用过量Driver DNA 目的是充分使Tester DNA 中和Driver DNA 序列相同的片段杂交,酶解去除,只有差异双链差异cDNA 经PCR几轮循环得以有效富集。
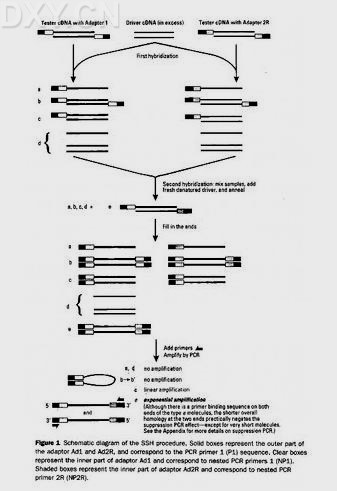
(二)抑制性扣除杂交(SSH)
如果两处理间存在较大差异,以及某些基因在Tester中存在上调表达(up-regulated expression)时,用RAD方法则达不到预期目的。于是,Diatchenko等于1996年提出了一种可以有效克服基因上调表达所造成不利后果的基因的新方法――抑制性扣除杂交(SSH)。该方法运用了杂交二级动力学原理,即丰度高的单链cDNA 在退火时产生同源杂交速度快于丰度低的单链cDNA ,从而使原来在丰度上有差别的单链cDNA 相对含量达到基本一致。而所谓抑制PCR,则是利用链内退火优先于链间退火,从而使非目的序列片段两端反向重复序列在退火时产生类似发卡的互补结构,无法与引物配对,从而选择性地抑制了非目的基因片段的扩增。
其具体过程为:
①与RAD相同,酶切后得到Tester与Driver样本的平末端cDNA 片段;
②将Tester cDNA 均分两份,分别接上接头1(adaptor 1)和接头2(adaptor 2)。接头设计上,adaptor由一长链(40 nt)和一短链(10 nt)组成的一端是平端的双链DNA 片段,长链3′端与cDNA 5′端相连。长链外侧序列(约20 nt)与第一次PCR引物序列相同,而内侧序列则与第二次引物序列同。此外,接头上含有T7启动子序列及内切酶识别位点,为以后连接载体和测序 提供方便;
③用过量的Driver样品分别与两份Tester样品进行第一次扣除杂交,分别得到4种产物。这种不充分杂交使得单链cDNA 分子在浓度上基本相同。同时由于Tester cDNA 中与Driver cDNA 序列相同片段大都形成异源双链分子,使得Tester cDNA 中差异表达基因得到第一次富集;
④混合两份杂交样品,同时加入新的变性Driver cDNA 进行第二次扣除杂交。此次杂交只有第一次杂交后经扣除和丰度均等化的单链Tester cDNA 能与Driver cDNA 形成双链分子。这一次杂交进一步富集了差异表达的cDNA ,并且还形成了两个5′端分别接不同接头的双链分子;
⑤5′填平末端,加入根据接头长链序列设计的内、外侧引物进行PCR扩增。第一次PCR是基于其抑制效应,只有两端分别是接头1和接头2的具差别表达的序列片段得以指数扩增。第二次PCR极大提高扩增的特异性,使得差异表达的目的基因片段得到大量富集,并可用于后续的筛选工作。
(三)代表性差示分析(RAD)和抑制性扣除杂交(SSH)优缺点分析
代表性差示分析(RAD)优点可以概括为以下几个方面:①引物设计十分巧妙;②可重复性好;③假阳性少;④mRNA 用量少、特异性高。其缺点为:①Tester组低丰度的分子自身杂交的几率很低,所以被扩增和的几率仍很低;②酶切连接步骤太多,cDNA 会损失很多,所以可能漏掉关键基因;③得到的不是cDNA 全长而是其酶切片段。
SSH自从在基因的实验中采用以来,其优势是非常明显的,主要表现在:①极大降低了假阳性率,由于SSH方法采用两次扣除杂交和两次PCR,保证了该方法具有较高特异性。据有关报道证明SSH方法假阳性率可降至6%;②具高度灵敏性,由于在反应中将丰度不同的单链分子含量归一化,保证了低丰度mRNA 检测成功;③SSH法在一次反应中,可同时分离出上百个差异表达的基因,这一点远胜于DDRT-PCR 和RAD。而其缺点与RAD类似。
四、交互扣除RNA 差别显示技术(RSDD)
交互扣除RNA 差别显示技术(RSDD)是由Kang等于1998年最新提出的结合扣除杂交和RNA 差别显示两项技术发展而出的一种有效、快速分离差别表达基因序列的方法。该方法应用于肿瘤进展的研究,可鉴定和已知的和高比率(>65%)的未知序列,这包括分别作为增强和抑制表达进展功能的cDNA s,分别称为增强进展基因(PEGen)和抑制进展基因(PSGen)。1999年通过交互扣除RNA 差别显示技术已鉴定出了16条差别表达的基因,其中有5条为未知的PEGen,6条为未知的PSGen。该方法主要由三部分组成:
①交互扣除(reciprocal subtraction):首先是要获得两个具有差别基因表达的细胞系的cDNA 文库,然后将这两个文库进行交互扣除杂交,从而获得两个扣除cDNA 文库。随后通过体内剪切得到质粒cDNA 文库;
②差异显示(differential display):由交互扣除cDNA 文库获得的纯化质粒可直接进行差异显示,这包括PCR扩增(加入3种3′锚定引物和18种5′随机引物)和进行5%序列胶电泳并切割回收差异显示条带;
③表达分析(expression analysis):运用反向Northern印迹分析(reverse Northern blotting)和Northern Blot分析鉴定经再扩增的DNA 片段的差异表达。反向Northern Blotting是将经差异显示得到的扩增后的DNA 片段转膜,并分别与来自两个不同细胞系总RNA 经反转录制备的32P标记的cDNA 第一链探针(reverse transcribed 32P-cDNA probe)进行杂交。234条再次扩增的DNA 片段中有181条被探测到(77%),其差异表达显示水平比两种细胞间Northern blot分析要高1.8倍。最后经Northern blotting分析得到两种细胞系差别表达的DNA 片段被到载体上进行测序 分析。RSDD目前被证实对于发生在复杂以及细胞生理变化中差异表达的基因有效且快速鉴定是很有价值的。
五、基因表达系列分析(SAGE)
基因表达系列分析(SAGE)是通过快速和详细分析成千上万个EST(express sequenced tags)来寻找出表达丰富度不同的SAGE标签序列。该法中,通过限制性酶切可以产生非常短的cDNA (10-14 bp)标签,并通过PCR扩增和连接,随后对连接物进行测序 。SAGE大大简化和加快了3’端表达序列标签的收集和测序 。同DD一样,SAGE是一个“开放”的系统,可以发现新的未知的序列。在进行样本的比较之前,SAGE在cDNA 的产生和处理上需要较多个步骤。由于SAGE是一个依赖DNA 测序 的基因计量方法,它对基因表达的测定比DD更加量化。由于需要进行大量的测序 反应,所以费用因素使大多数研究机构对其广泛应用的主要限制。
六、电子消减
电子消减是通过比较相同的基因的cDNA 文库中被重复测序 出现次数,可以粗略推算出该基因在不同文库中的丰度差异以达到差异显示的目的。
七、差异显示技术衍生的方法
(一)应用随机引物PCR的RNA 指纹法
除反转录这步是由随机oligo dT引物替代锚定oligo dT引物外,应用随机引物PCR的指纹法(RNA fingerprinting by arbitrarily primed PCR,RAP-PCR)与DD-PCR基本上相同。与DD-RCR相似,RAP-PCR可以用于多个不同细胞族或不同处理后转录体之间的比较。
(二)引物选择差异显示
引物选择差异显示(DD-PCR with selected primers,SPR)的目的是进一步倾向于筛选低到中丰富度RNA 。与RAP和DD-PCR不同,SPR实验性的选择随即引物以避免高丰富度RNA 的扩增。特点有4个方面:①所设计引物因含有50%的G+C,但3′末端应不超过50%,以减少高丰度rRNA 和线粒体RNA 的扩增。②所有PCR循环应以较高的退火温度进行,以增加选择性和可重复性,减少所扩增条带的数目。③由于高丰度转录体有一个引物优先扩增,条带的选择将根据两个引物的扩增为依据。④由于Northern分析不能检验地丰富度的RNA ,差异表达条带的鉴定将由定量PCR来确定。这些改进也在其他研究中采用。SPR这一方法的缺点是引物必须在实验中设计。然而,一旦选择了这一引物,就可以在类似实验中使用。
(三)目标显示
DD-PCR一个非常有用的改进是目标显示(targeted display)。它可以鉴定基因家族的成员、具有特殊区域的基因或不同条件下分离的序列图。目标显示用锚定引物进行反转录,而PCR扩增则采用与保守的蛋白区域同源的变性引物来进行。正常情况下,基因家族新成员可以通过用变性寡核苷酸率选基因文库来进行鉴定。近来,用两个变性引物进行PCR的方法也已使用。此方法的主要缺陷是:①5′段引物应具有与标定区域100%同源的序列;②引物不能过度变性,以至产生过度复杂模式;③目标区域应位于近poly A尾处。
(四)顺序差异显示
顺序差异显示(ordered differential display,READS)首先由Prashar和Weissman报道。其反转录由一个含有20个碱基尾端(heel)的双核苷酸oligo dT来进行,因此所有cDNA 均有共同的3’端。这些产物用限制性酶消化并连接到一个Y型的连接体(adapter)。Y型的连接体有三部分组成:①与限制酶相容的3′端开口;②补充的中间体;③非补充5′。在扩增中,3′引物延伸到“跟部”,而5′端引物延伸到Y型连接体的“上肢”。但只有限制消化的cDNA 的3′端在严紧条件下扩增。READS的结果依赖于消化cDNA 的限制酶,因此cDNA 的长度各不相同。不同细胞总RNA 获得的cDNA 由不同的限制酶可以系统的分切为3′端限制性片断。用限制性酶切来消化cDNA ,几乎可以反映所有哺乳动物的。其两次实验间的重复性很好。
八、基因技术
生物芯片技术是在信息技术和生物技术集合在一起的产物。其中的基因借助于计算机控制的机械手段对已知的众多探针按预定的位置点阵,固定在固相支持片基上,然后于标记的待测样品进行分子杂交反应,通过激光共聚焦荧光检测系统来分子每个杂交信号的有无及强弱,进而获得每个样品携带的相关信息,经专用的程序软件的分析得出最终结论。该方法可以一次对样品中的大量相关信息进行平行监测分析,解决了传统核算杂交、免疫反应的操作复杂、检测效率低既可比性不强等问题。该方法的最基本原理也是基于基因的差别表达,因现阶段常将其作为功能学的研究的重要方法。