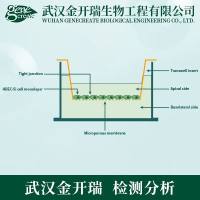
蛋白表达及纯化研究中的FAQ,转自生物通
丁香园论坛
2075
一、原核蛋白表达及纯化
1、怎样提高E.coli中6′His标签蛋白的表达水平?
6 × His标签蛋白的表达水平低原因可能为:目标蛋白有毒或不稳定,或者是细菌生长过程中不能维持表达结构。有时候,插入片段的5’端序列编码了干扰转录或翻译的元件(比如有反向重复序列而形成茎环结构,掩盖了Shine-Dalgarno序列),在这种情况下,有必要检查和修改表达序列。改变培养基和宿主菌也可能会有所帮助。
外源性蛋白的表达会使宿主菌生长迟缓。高速的转录、翻译重组蛋白所需要的代谢消耗,导致宿主菌生长的缓慢。低浓度下即影响宿主菌生长的基因产物被认为‘有毒’,这样的产物包括细胞膜蛋白或与DNA相互作用或干扰电子传递的蛋白。为了降低有毒蛋白在被诱导前对宿主菌生长的影响,在无诱导的时候的基础转录水平越低越好,并且诱导前的细菌传代数要控制在最小范围。细菌生长过程中质粒的丢失有时候可以通过在培养基中加入高浓度的ampicillin (200ug/ml) 或carbenicillin (50ug/ml)得到解决。对于一些不稳定的表达结构,应当避免过夜培养起始菌。先从新鲜的培养基上挑选单克隆,少量培养后,2000倍稀释大量培养。
含有疏水基团的重组蛋白往往对宿主菌有毒性。因此,除非有特殊的意义,应当去除插入片段中编码信号肽序列或穿膜区的序列。然而,也有少数包含信号肽的蛋白可以在膜间质少量的表达。这种情况下,建议诱导前培养温度降低为25度。
有些蛋白,尤其是小于10KDa的蛋白在E. coli中不稳定,可能很快被细菌中的蛋白酶降解。克服这种困难的方法包括:降低培养温度到30度;使用一种或多种蛋白酶缺陷的宿主菌;使用QIAGEN公司的表达质粒pQE-16、pQE-40,使生成目标多肽与DHFR的融合蛋白;如果是纯化过程中蛋白易降解,则可以在纯化试剂中加入甘油以及蛋白酶抑制剂,并且确保整个操作在4度完成。
2、如何在E. coli中表达高度毒性的蛋白?
在E. coli中表达高度毒性的蛋白往往是很困难的。毒性基因表达‘泄漏’会杀死宿主菌,尤其在早期的生长或转化后,可以使含有突变的不表达质粒的细菌选择性生长。因此,要表达高毒性的蛋白,需要更高水平的lac 抑制蛋白。推荐同时使用QIAGEN公司的pQE-80L系列表达载体和M15[pREP4]宿主菌。pQE-80L系列表达载体带有lac抑制子基因,宿主菌M15[pREP4]带有pREP4质粒,两者持续的正向或反向高水平表达lac 抑制蛋白。这样协同作用,可以使诱导前的表达降低到最小的水平,因而提高成功表达毒性蛋白的可能性。
3、如何提高E. coli中可溶性蛋白的表达量?
在E. coli 中表达的真核细胞蛋白往往形成不可溶的包含体。原因可能包括疏水基团的相互作用,半胱氨酸在E. coli 胞质还原性环境中不能正确形成二硫键。
QIAexpress 纯化系统的一个优点就在于包含体中带有6 × His标签的蛋白可以在变性剂作用下生成可溶性蛋白,然后用Ni-NTA纯化。如有必要,变性条件下纯化的蛋白可以复性,重新折叠。
尽管如此,很多研究者还是发现纯化过程中的复性比较困难。另一个方法是调整表达环境使生成少量的可溶的天然构象的重组蛋白。即使形成了包含体,一些带6×His标签的蛋白仍可以在胞质中,这些胞质中的可溶性蛋白可以在天然的非变性的条件下被纯化。如果需要更多的可溶性蛋白,诱导后降低培养温度会有所帮助。培养温度常常直接影响蛋白表达水平和可溶性,降低温度可以降低表达水平从而使可溶性蛋白量增加。另外,也可以在诱导前让培养物达到更高的细胞密度而表达期控制在最短时间。IPTG的浓度可以从1mM降低到0.0005mM,使表达水平降低90%以上。并且,改变宿主菌也可能有效,因为某些菌比其它菌对某些蛋白的耐受性更好,可以生成更高浓度的可溶性蛋白。最后,很多蛋白需要金属协同因子才能够保持其可溶状态,因此在培养物中加入金属盐可能会有帮助。如果不知道蛋白所需的金属协同因子,则有必要测试不同的添加物。需要注意的是一些二价的阳离子会影响蛋白与Ni-NTA的结合。
4、如何在使用Ni-NTA纯化蛋白过程中去除富含组氨酸的杂蛋白?
在结合试剂与洗脱试剂中加入20mM的咪唑(即使在变性条件下),可以抑制杂蛋白的结合。实验中逐步提高咪唑的浓度,以确定最佳的去除杂蛋白
的浓度。
过量使用Ni-NTA也会导致非特异性的结合。因此,对应于预期的表达蛋白量来确定合适的结合体系,可以减少非特异蛋白的结合。
很多杂蛋白可能是由于非特异的离子作用引起的,因此纯化过程中确保在试剂中加入至少300mM的NaCl。
杂蛋白也可能与标签蛋白结合。加入b-ME(到20mM)可以解离杂蛋白与6′His标签蛋白间形成的二硫键。这在目标蛋白中包含相对多的半胱氨酸时尤其重要。
杂蛋白与6′His标签蛋白的非特异性结合也可能是通过疏水作用产生。在洗脱液中加入以下物质可以减少因此而产生的非特异性:非离子洗涤剂(Trion X-100或Tween 20);30%的乙醇或甘油。注意,不同的蛋白纯化所需加入的最佳浓度不同,需要试验摸索。
杂蛋白也可能是截断的标签蛋白。这样的杂蛋白可以用Western Blot技术很容易的识别。通过质粒测序,检查出内部的翻译起始位点或成熟前的终止子。
5、Ni-NTA 是否能用于纯化含有内部His标签的蛋白?
是的。Ni-NTA可以与His标签结合,无论标签位于蛋白的N端、C端或是内部。需要注意的是,在非变性条件下纯化时,His标签必须充分暴露在蛋白表面以利于充分的纯化。
6、是否可以把His标签从表达蛋白上切割掉?
是的。QIAGEN提供一种载体,称为pQE-30 Xa, 与pQE-30相似,但在His标签与多克隆位点之间有编码因子Xa蛋白酶识别位点的序列。因子Xa蛋白酶可以在蛋白酶识别位点的精氨酸之后进行切割。经过因子Xa蛋白酶处理可以得到不含组氨酸的重组蛋白。
7、哪一种抗His抗体最灵敏?
Penta His和Tetra His 抗体可以与含有6′His标签的蛋白结合,无论标签附近的氨基酸组成如何,甚至是在标签部分掩盖的情况下。这些抗体相对应的抗原识别位点的微小区别使得某些含有6′His标签的蛋白对某一个抗体的灵敏度高与另一个抗体。因此,为了得到最佳的结果,建议在实验的开始平行比较这些抗体。QIAGEN为此提供一种试剂盒Antibody Selector Kit,用于选择最佳的抗体。6His Protein Ladder 提供了蛋白分子量标准和Western Blot的阳性参照。
二、真核荧光蛋白的表达
1、 检测不到瞬时转染细胞中的GFP表达,该怎么办?
1) 确定检测的方法正确,例如滤波片的设置;
2) 如果转染的是重组质粒,先用空白的质粒(无插入片段)作为对照,以确定检测方法正确;
3) 如果是重组质粒,对质粒与插入片段的连接处测序,确定阅读框正确;
4) 换一种转染细胞,常用的细胞有HeLa, COS, HEK293, CHO-K1, NIH3T3;
5) 优化转染条件,例如质粒量、转染试剂、转染时的细胞丰度。
2、 挑选了稳定表达GFP的克隆,培养后,发现荧光变弱或是不稳定,可能的原因是什么?
筛选任何稳定表达的基因,都有可能遇到类似的问题,因为荧光检测特别敏感、方便,所以这类问题更容易被观察到。出现这类问题,可能的原因是:
1) 挑选的单克隆不纯,混有其他细胞;
2) 即使是单克隆来源的细胞群,荧光的表达也可能不同。选择压力、细胞的生长丰度的改变都可能引起荧光表达不同。
3、挑选了稳定表达GFP的克隆,培养后,发现荧光变弱或是不稳定,该怎么办?
1) 瞬时转染时,质粒进入细胞后以游离的形式存在。目的基因的表达是通过细胞的转录与翻译机制进行的。一个细胞可能‘吞入’数个质粒,因此细胞间会存在差异。总的来说,稳定转染的细胞只含有1-2个被整合的质粒。因此,稳定转染细胞的荧光表达量会低于瞬时转染的细胞;
2) 确定细胞转染48小时后或是新霉素筛选前才长满。因为生长接触抑制会降低目的基因的表达;
3)确定细胞在转染后48小时以及药物筛选过程中生长状态良好。如果在瞬时表达阶段细胞不健康,可能是提示有潜在的毒性;
4)做G418杀伤曲线。如果筛选用的药物太浓(不同批号间的药物浓度可能不同),选择压力过强。报告基因的表达水平与筛选抗性基因的不完全正相关,细胞在过高浓度的药物中生长,报告基因的表达反而降低。过高的药物浓度也可能影响了细胞的生长状态,进而影响报告基因的表达。这些副作用在长时间的筛选过程中尤其明显。筛选后的细胞培养,建议药物浓度降低为筛选时的1/2到1/5;
5)总的来说,细胞培养时间在1-2个月内表达比较稳定,因此,建议分装冻存筛选过后的细胞,然后复苏原管,以确保细胞的统一性;
6)可以通过流式细胞仪分析筛选高表达的细胞,以得到高水平表达的细胞株。这一筛选过程可以在药物筛选完成之后进行。
1、怎样提高E.coli中6′His标签蛋白的表达水平?
6 × His标签蛋白的表达水平低原因可能为:目标蛋白有毒或不稳定,或者是细菌生长过程中不能维持表达结构。有时候,插入片段的5’端序列编码了干扰转录或翻译的元件(比如有反向重复序列而形成茎环结构,掩盖了Shine-Dalgarno序列),在这种情况下,有必要检查和修改表达序列。改变培养基和宿主菌也可能会有所帮助。
外源性蛋白的表达会使宿主菌生长迟缓。高速的转录、翻译重组蛋白所需要的代谢消耗,导致宿主菌生长的缓慢。低浓度下即影响宿主菌生长的基因产物被认为‘有毒’,这样的产物包括细胞膜蛋白或与DNA相互作用或干扰电子传递的蛋白。为了降低有毒蛋白在被诱导前对宿主菌生长的影响,在无诱导的时候的基础转录水平越低越好,并且诱导前的细菌传代数要控制在最小范围。细菌生长过程中质粒的丢失有时候可以通过在培养基中加入高浓度的ampicillin (200ug/ml) 或carbenicillin (50ug/ml)得到解决。对于一些不稳定的表达结构,应当避免过夜培养起始菌。先从新鲜的培养基上挑选单克隆,少量培养后,2000倍稀释大量培养。
含有疏水基团的重组蛋白往往对宿主菌有毒性。因此,除非有特殊的意义,应当去除插入片段中编码信号肽序列或穿膜区的序列。然而,也有少数包含信号肽的蛋白可以在膜间质少量的表达。这种情况下,建议诱导前培养温度降低为25度。
有些蛋白,尤其是小于10KDa的蛋白在E. coli中不稳定,可能很快被细菌中的蛋白酶降解。克服这种困难的方法包括:降低培养温度到30度;使用一种或多种蛋白酶缺陷的宿主菌;使用QIAGEN公司的表达质粒pQE-16、pQE-40,使生成目标多肽与DHFR的融合蛋白;如果是纯化过程中蛋白易降解,则可以在纯化试剂中加入甘油以及蛋白酶抑制剂,并且确保整个操作在4度完成。
2、如何在E. coli中表达高度毒性的蛋白?
在E. coli中表达高度毒性的蛋白往往是很困难的。毒性基因表达‘泄漏’会杀死宿主菌,尤其在早期的生长或转化后,可以使含有突变的不表达质粒的细菌选择性生长。因此,要表达高毒性的蛋白,需要更高水平的lac 抑制蛋白。推荐同时使用QIAGEN公司的pQE-80L系列表达载体和M15[pREP4]宿主菌。pQE-80L系列表达载体带有lac抑制子基因,宿主菌M15[pREP4]带有pREP4质粒,两者持续的正向或反向高水平表达lac 抑制蛋白。这样协同作用,可以使诱导前的表达降低到最小的水平,因而提高成功表达毒性蛋白的可能性。
3、如何提高E. coli中可溶性蛋白的表达量?
在E. coli 中表达的真核细胞蛋白往往形成不可溶的包含体。原因可能包括疏水基团的相互作用,半胱氨酸在E. coli 胞质还原性环境中不能正确形成二硫键。
QIAexpress 纯化系统的一个优点就在于包含体中带有6 × His标签的蛋白可以在变性剂作用下生成可溶性蛋白,然后用Ni-NTA纯化。如有必要,变性条件下纯化的蛋白可以复性,重新折叠。
尽管如此,很多研究者还是发现纯化过程中的复性比较困难。另一个方法是调整表达环境使生成少量的可溶的天然构象的重组蛋白。即使形成了包含体,一些带6×His标签的蛋白仍可以在胞质中,这些胞质中的可溶性蛋白可以在天然的非变性的条件下被纯化。如果需要更多的可溶性蛋白,诱导后降低培养温度会有所帮助。培养温度常常直接影响蛋白表达水平和可溶性,降低温度可以降低表达水平从而使可溶性蛋白量增加。另外,也可以在诱导前让培养物达到更高的细胞密度而表达期控制在最短时间。IPTG的浓度可以从1mM降低到0.0005mM,使表达水平降低90%以上。并且,改变宿主菌也可能有效,因为某些菌比其它菌对某些蛋白的耐受性更好,可以生成更高浓度的可溶性蛋白。最后,很多蛋白需要金属协同因子才能够保持其可溶状态,因此在培养物中加入金属盐可能会有帮助。如果不知道蛋白所需的金属协同因子,则有必要测试不同的添加物。需要注意的是一些二价的阳离子会影响蛋白与Ni-NTA的结合。
4、如何在使用Ni-NTA纯化蛋白过程中去除富含组氨酸的杂蛋白?
在结合试剂与洗脱试剂中加入20mM的咪唑(即使在变性条件下),可以抑制杂蛋白的结合。实验中逐步提高咪唑的浓度,以确定最佳的去除杂蛋白
的浓度。
过量使用Ni-NTA也会导致非特异性的结合。因此,对应于预期的表达蛋白量来确定合适的结合体系,可以减少非特异蛋白的结合。
很多杂蛋白可能是由于非特异的离子作用引起的,因此纯化过程中确保在试剂中加入至少300mM的NaCl。
杂蛋白也可能与标签蛋白结合。加入b-ME(到20mM)可以解离杂蛋白与6′His标签蛋白间形成的二硫键。这在目标蛋白中包含相对多的半胱氨酸时尤其重要。
杂蛋白与6′His标签蛋白的非特异性结合也可能是通过疏水作用产生。在洗脱液中加入以下物质可以减少因此而产生的非特异性:非离子洗涤剂(Trion X-100或Tween 20);30%的乙醇或甘油。注意,不同的蛋白纯化所需加入的最佳浓度不同,需要试验摸索。
杂蛋白也可能是截断的标签蛋白。这样的杂蛋白可以用Western Blot技术很容易的识别。通过质粒测序,检查出内部的翻译起始位点或成熟前的终止子。
5、Ni-NTA 是否能用于纯化含有内部His标签的蛋白?
是的。Ni-NTA可以与His标签结合,无论标签位于蛋白的N端、C端或是内部。需要注意的是,在非变性条件下纯化时,His标签必须充分暴露在蛋白表面以利于充分的纯化。
6、是否可以把His标签从表达蛋白上切割掉?
是的。QIAGEN提供一种载体,称为pQE-30 Xa, 与pQE-30相似,但在His标签与多克隆位点之间有编码因子Xa蛋白酶识别位点的序列。因子Xa蛋白酶可以在蛋白酶识别位点的精氨酸之后进行切割。经过因子Xa蛋白酶处理可以得到不含组氨酸的重组蛋白。
7、哪一种抗His抗体最灵敏?
Penta His和Tetra His 抗体可以与含有6′His标签的蛋白结合,无论标签附近的氨基酸组成如何,甚至是在标签部分掩盖的情况下。这些抗体相对应的抗原识别位点的微小区别使得某些含有6′His标签的蛋白对某一个抗体的灵敏度高与另一个抗体。因此,为了得到最佳的结果,建议在实验的开始平行比较这些抗体。QIAGEN为此提供一种试剂盒Antibody Selector Kit,用于选择最佳的抗体。6His Protein Ladder 提供了蛋白分子量标准和Western Blot的阳性参照。
二、真核荧光蛋白的表达
1、 检测不到瞬时转染细胞中的GFP表达,该怎么办?
1) 确定检测的方法正确,例如滤波片的设置;
2) 如果转染的是重组质粒,先用空白的质粒(无插入片段)作为对照,以确定检测方法正确;
3) 如果是重组质粒,对质粒与插入片段的连接处测序,确定阅读框正确;
4) 换一种转染细胞,常用的细胞有HeLa, COS, HEK293, CHO-K1, NIH3T3;
5) 优化转染条件,例如质粒量、转染试剂、转染时的细胞丰度。
2、 挑选了稳定表达GFP的克隆,培养后,发现荧光变弱或是不稳定,可能的原因是什么?
筛选任何稳定表达的基因,都有可能遇到类似的问题,因为荧光检测特别敏感、方便,所以这类问题更容易被观察到。出现这类问题,可能的原因是:
1) 挑选的单克隆不纯,混有其他细胞;
2) 即使是单克隆来源的细胞群,荧光的表达也可能不同。选择压力、细胞的生长丰度的改变都可能引起荧光表达不同。
3、挑选了稳定表达GFP的克隆,培养后,发现荧光变弱或是不稳定,该怎么办?
1) 瞬时转染时,质粒进入细胞后以游离的形式存在。目的基因的表达是通过细胞的转录与翻译机制进行的。一个细胞可能‘吞入’数个质粒,因此细胞间会存在差异。总的来说,稳定转染的细胞只含有1-2个被整合的质粒。因此,稳定转染细胞的荧光表达量会低于瞬时转染的细胞;
2) 确定细胞转染48小时后或是新霉素筛选前才长满。因为生长接触抑制会降低目的基因的表达;
3)确定细胞在转染后48小时以及药物筛选过程中生长状态良好。如果在瞬时表达阶段细胞不健康,可能是提示有潜在的毒性;
4)做G418杀伤曲线。如果筛选用的药物太浓(不同批号间的药物浓度可能不同),选择压力过强。报告基因的表达水平与筛选抗性基因的不完全正相关,细胞在过高浓度的药物中生长,报告基因的表达反而降低。过高的药物浓度也可能影响了细胞的生长状态,进而影响报告基因的表达。这些副作用在长时间的筛选过程中尤其明显。筛选后的细胞培养,建议药物浓度降低为筛选时的1/2到1/5;
5)总的来说,细胞培养时间在1-2个月内表达比较稳定,因此,建议分装冻存筛选过后的细胞,然后复苏原管,以确保细胞的统一性;
6)可以通过流式细胞仪分析筛选高表达的细胞,以得到高水平表达的细胞株。这一筛选过程可以在药物筛选完成之后进行。