【资源】大规模蛋白质相互作用研究方法进展
丁香园论坛
2368
摘 要:随着2000 年酵母大规模蛋白质相互作用网络图谱的成功描绘,蛋白质相互作用特别是大规模蛋白质相互作用研究成为生命科学领域的又一个研究热点。酵母、果蝇、线虫以及人类蛋白的大规模相互作用图谱的相继完成,不仅对系统研究细胞内各种生命活动有着重要意义,也标志着蛋白质相互作用研究方法的不断发展和完善。本文综述了当前大规模研究蛋白质相互作用的技术方法并进行了比较分析。每种技术都有其各自的优缺点,在实验中要根据不同的要求和目的选择适宜的方法。
关键词:蛋白质相互作用;酵母双杂交;串联亲和纯化
The advance in research methods for large-scale protein-protein interactions
GUAN Wei, WANG Jian, HE Fu-Chundefined
(Beijing Institute of Radiation Medicine, Beijing 100850, China)
Abstract: In 2000, the first network of large-scale protein-protein
interactions was accomplished in yeast. It was a historic moment and became
another hot spot of protein research. Subsequently, such proteins interaction
maps were also brought to success in the fly, worm and even human during five
years. So rapid progress in large-scale protein-protein interactions turned up
many new angles of view for investigating the mechanisms of cell functions,
actually it also suggested the development and complement of research method
itself. In this article we summarized the progress of techniques in
large-scale protein-protein interactions and showed their differences. Anyway,
the researchers should choose right means to perform their diverse intentions.
Key words: protein-protein interaction; yeast two hybrid; tandem affinity
purification
人类基因组测序的完成标志着一个新的生物学研究时代——后基因组时代(post-genomic era)的来临。科学家们的研究热点又回到蛋白质上,全基因组的序列信息并不足以解释及推测细胞的各种生命现象,蛋白质才是细胞活性及功能的最终执行者。构成细胞的所有组成蛋白有多少;他们的动态相互作用关系又如何;细胞内所有蛋白即蛋白质组又是怎样组织在一起形成有功能、有秩序的网络结构?
这些问题的解决才是最终阐明细胞中各种生命活动机制的关键所在[1]。
1 大规模蛋白质相互作用研究的目的及意义
生物体内各种生命信息由不同的基因经转录、翻译传递到相应的蛋白质上并使其具有各自的生化特性及生物学活性;但每个蛋白质并不是独立的在细胞中完成被赋予的功能,它们在细胞中通常与其他蛋白质相互作用形成大的复合体,在特定的时间和空间内完成特定的功能,而且有些蛋白质的功能只有在复合体形成后才能发挥出来,如依赖于构象变化或翻译后修饰的蛋白质功能[2]
;另一方面,某些蛋白质可能参与了不止一个的复合体,简单的两两相互作用研究就不足以阐明这种更为复杂的相互作用,因此,大规模、高通量的蛋白质相互作用研究应运而生,其目的是在细胞的特定生理条件下,从一个蛋白到多个蛋白,从一个复合体到多个复合体,进而描绘出整个蛋白质组中蛋白间相互作用的网络图。基于这些作用关系,科学家们才能从真正意义上阐明一个蛋白质的功能,才有可能研究细胞中某一生理活动中所有相关蛋白质的变化及作用机制。大规模蛋白质相互作用的研究还有助于了解细胞中不同生命活动之间的相互关系。
2 大规模蛋白质相互作用的研究策略及方法
研究蛋白质相互作用时要根据不同的实验目的及条件选择不同的实施策略。研究已知蛋白间的相互作用人们关注的是蛋白间能否发生结合,实验本身更趋向于验证性,因此,应选择操作性强、可信度高、接近生理条件的技术方法,尽量减少实验本身带来的假阴性或假阳性。若研究者想得到细胞中能与某一蛋白结合的所有未知蛋白质,则实验的关键是得到细胞中含有靶蛋白的复合体,经过有效地分离纯化后,逐一鉴定复合物中的各个组分;或者采用一对多的筛选方法,如酵母双杂交,在建立的蛋白表达文库中检测可能与靶蛋白相互作用的蛋白质。值得注意的是在这一策略中实验结果的进一步验证是必不可少的。目前用于大规模研究蛋白质间相互作用的方法包括酵母双杂交、串联亲和纯化、质谱鉴定、蛋白芯片以及基于生物信息学的分析方法等。
2.1 酵母双杂交 该系统由Fields 和Song[3]首先在研究真核基因转录调控时建立。利用真核生长转录因子的两个不同的结构域:DNA结合结构域(DNAbinding domain)和转录激活结构域(transcription-activating domain),分别与目标蛋白X 及可能与目标蛋白相互作用的蛋白Y 相连,并共同转入酵母细胞。如果蛋白X与Y能够发生相互作用就能使转录因子原来分开的两部分结合,形成完整的活性形式从而激活下游报告基因。通过检测报告基因的表达产物就可判断两种蛋白是否发生相互作用[4~5]。
这一经典的蛋白相互作用研究方法接近于体内环境,那些瞬时、不稳定的两两相互作用也可以被检测到,并且与内源蛋白的表达无关[6]。鉴于这些优点并结合简便高效的Gateway表达载体构建方法[7],在大规模的蛋白质-蛋白质相互作用研究中酵母双杂交系统得到了最为广泛的应用。Rain 等[8]用该法绘制了人类胃肠道病原菌Helicobacter pylori 的大规模蛋白质相互作用图谱。在261 种蛋白中,确立了 1200 种相互作用关系,涵盖了整个蛋白质组得46. 6%。随后,Giot 等[9]和Li 等[10]在果蝇和线虫中也成功地研究了大规模的蛋白质相互作用。除了对模式生物的研究外,Stelzl 等[11]和Raul 等[12]则先后分析了人脑组织、人已知ORF中大规模的蛋白质相互作用网络。但酵母双杂交方法本身也有一定的局限性:(1)不能研究具有自激活特性的蛋白质;(2)只能检测两个蛋白间的相互作用;(3)检测的相互作用需发生在细胞核内,对于不能定位到细胞核中的蛋白质无法研究;(4)大部分实验中有将近50%的假阳性率,且推测的相互作用仅有3% 在两种以上的实验中得到验证。为了弥补方法本身的缺点及局限性,研究者也不断地对其进行完善和改进。Stelzl 等[11]在研究中就采用了以下的策略:(1)选择不同功能、不同大小的蛋白作为诱饵,以确保所选靶蛋白在整个蛋白质组中的代表性;(2)筛选过程中采用两轮杂交的方法:第一轮以混合诱饵(8 个)对文库进行筛选,结果呈阳性的克隆再进行一对一的第二轮杂交,这样既降低了工作量又提高了结果的准确性;(3)pull-down,免疫共沉淀对酵母双杂交的结果进行体内的相互作用验证;(4)利用生物信息学的方法对结果进行系统分析,包括基因的染色体定位、蛋白质作用网络的拓扑结构分析等,从多方面分析结果的可信度。据此,他们最终确认了911 对高可信度的相互作用涉及到401 种蛋白,数据分析中设立了6 个标准来判定得到的结果,大大提高了酵母双杂交实验结果的可信度。
2.2 串联亲和纯化 Rigaut 等[13]首次采用串联亲和纯化技术(tandem affinity
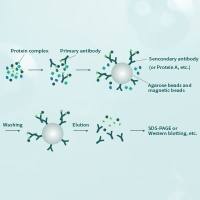
purification,TAP)研究了蛋白间的相互作用,与其他融合标签方法的最大不同是该方法选用了两个连续的标签而不是通常意义上的一个。标签共分三部分:蛋白A 、C B P (calmodulin binding peptide,钙调素结合多肽)和中间连接的TEV 酶识别的酶切位点。带有TAP 标签的融合蛋白在细胞中表达与内源相互作用蛋白形成复合物,细胞裂解后经IgG 偶联的层析柱纯化,蛋白A 与IgG 特异结合,从而使含有标签的复合物得到第一次纯化。为了除去与柱填料非特异结合的蛋白,用TEV 酶切割分离蛋白A 标签,使含有靶蛋白的复合物与层析柱分离并经过钙调素偶联的亲和层析柱进行第二次纯化,靶蛋白上的CBP标签可与钙调素结合;在加入过量螯合剂EGTA后CBP 与亲和层析柱分离使含有靶蛋白的复合体得到分离纯化, 经SDS-PAGE,复合物中的各个蛋白被分开,切胶、胰酶消化后,即可通过质谱鉴定复合物中各个蛋白的氨基酸序列(图1)。该方法的优点:(1)不需要过多的背景知识就可以得到大量含靶蛋白的复合体;(2)蛋白表达及与复合物的结合都接近生理水平,是一种检测体内蛋白相互作用的方法;(3)TAP 采用两步亲和纯化,提高了纯化产物的特异性。一般在2L 的酵母培养基中(细胞湿重约10g)可以分离纯化得到足够一维电泳及质谱分析的蛋白复合物[ 1 5 ] 。Gavin 等 [16]采用TAP 结合质谱鉴定,对啤酒酵母的多蛋白复合物进行了大规模的研究。共分析了 1 739 个基因其中与人类基因相关的有1 143 个,纯化了589 种蛋白,经生物信息学分析,它们分属于 232 个不同的多蛋白复合体;在细胞中具有新功能的为344 个,231 个蛋白的功能以前未见报道。由于TAP/MS方法的高灵敏性使得仅有15个拷贝的蛋白也能被检测到,鉴定的蛋白分子量从6.6kDa 到 559kDa,等电点从3.9 到12.4。这一研究提供了真核生物中二元以上的蛋白相互作用网络,从中得到的有关其功能的各种信息也能为药物的筛选提供依据。TAP 技术的出现有效地促进了酵母中大规模的蛋白鉴定和纯化工作,但是其在高等真核生物中的应用却处于滞后状态,这是因为在真核细胞中表达的内源靶蛋白与带有标签的蛋白会发生竞争作用,影响相互作用的研究。因此,Forler 等[17]将RNAi 与TAP 技术联用在果蝇的S2 细胞(Drosoph ila melanogaster)中研究了蛋白质间的相互作用,该策略避免了内源蛋白的干扰,进而从高等真核细胞中分离并鉴定了相应的蛋白复合体,实验表明,蛋白纯化的特异性及有效性均得到提高,采用这种方法他们还对靶蛋白生物功能进行了分析。iTAP(TAP 与RNAi)保持了TAP 方法在原核生物中应用的一些优势:(1)基于标签的亲和纯化方法;(2)用TAP 标记的蛋白替代内源基因表达产物;但由基因本身的启动子调控蛋白的表达量,这一点在真核细胞中还无法实现。对于这部分相互作用的研究,人们多采用逆转录病毒载体,将带有标签的靶蛋白引入细胞中。由于逆转录病毒载体能够稳定的整合在宿主细胞的基因组中并且拷贝量较低,因此,外源蛋白可以近似于生理水平的低表达,进而避免由于高表达而出现的假阳性结果。此外Zhou等[18]在原代细胞中做了有益的尝试,将TAP标签通过基因敲入技术引入小鼠中,使得该技术能够应用于更多的基因并进行组织或细胞类型特异的复合体研究。
图1 TAP亲和层析纯化原理[14]
注:A:标签中的ProteinA 与固化的IgG 结合缓冲液淋洗去除不能结合的杂蛋白,TEV 酶切分离ProteinA
和靶蛋白;B:利用CBP(Calmodulin binding peptide)-Calmodulin
的相互作用进一步纯化复合物,缓冲液洗去杂蛋白。加入过量的螯合剂(EGTA)螯合Ca2+,使纯化的复合物与层析柱分离。
TAP 纯化时存在蛋白污染问题,很难判断这些蛋白是真正的相互作用,还是蛋白处理过程中的副产品。为此,有人建议用三标签的方法[19],但实验表明该法也不能排除蛋白的污染。其他标签也有应用,比较流行的是Flag[20]。GST、His6、生物素等也分别用于蛋白纯化,但由于他们有限的亲和力及较高的背景,在大规模的蛋白质相互作用研究中未得到广泛应用。选择何种标签也许是个难题,每种标签都有自身的弱点:大标签适于规模较大的相互作用研究,但可能影响蛋白的正常折叠和功能;小标签可能会更大程度的保持蛋白本身的特性,但在表达纯化时通常含量较低[21]
2.3 质谱在蛋白质相互作用分析中的应用 在蛋白质相互作用的研究中,质谱最初只是作为鉴定蛋白质的一种快速、高效的方法:混合的蛋白样品首先经过纯化分离、然后通过SDS-PAGE
将复合体中的组分分开,随后对感兴趣的蛋白进行酶解;得到的肽段在质谱中进行鉴定,从而确定样品中的目标蛋白;但由于分离方法的灵敏度及技术本身的局限,对于那些相互作用较弱、分子量较大或偏碱性的蛋白往往不能达到理想的分析效果,增加了实验结果的假阴性。定量蛋白质组技术的出现解决了这一问题,而该技术的策略正是较好的利用了质谱的高灵敏度。目前已有许多种研究蛋白相互作用的定量蛋白质组学方法,以下就稳定同位素标记方法为例进行说明。稳定的同位素之间具有“轻”、“重” 之分,而其化学性质又保持相同,这就使得有同位素标记的蛋白在分离纯化时具有相同的特性,而在随后的质谱鉴定中又能根据质量数的差异被区分开。通常采用的同位素标记为2H、13C、15N 以及 18O。加入同位素标记的方法主要分为:代谢标记法(stable isotope-labelled animo acids in cell culture, SILAC)和化学标记法(isotope-coded affinity tagging, ICAT)。前者是在培养细胞的过程中加入同位素标记(体内标记) ;后者则是在提取细胞内总蛋白并经过亲和纯化后加入同位素标记(体外标记)。最后两种方法都将得到的对照及实验组样品混合,进行质谱分析。由于同位素“ 轻”、“重” 差异, 质谱能很容易的区分并鉴定实验组中特异的蛋白质。该方法中,亲和纯化过程是最大限度的保留实验组中的差异蛋白而非排除污染蛋白,系统本身的灵敏度在质谱分析中得到保证,这就避免了传统的定性蛋白质组技术中分离方法本身固有的局限性,大大提高了实验结果的灵敏度和准确性。Blagoev等[22]采用SILAC 策略研究了在EGF 刺激细胞后,能够与 Grb2 的SH2结构域结合的相互作用蛋白。两组细胞分别在含有不同同位素(12C-Arg,13C-Arg)的培养基中生长一段时间,EGF 刺激其中一组细胞(实验组)10 分钟,随后两组细胞同时裂解、纯化并进行LC-MS/ MS 分析。最终228 个蛋白得到鉴定,其中28 个蛋白在被刺激的实验组中表达水平明显高于对照组。这些蛋白除了已知的信号分子外,还有一些蛋白参与了信号衰减或具有细胞骨架的功能,此外还鉴定到了未知蛋白质。这表明该方法在研究信号通路中具有较大的潜力,值得一提的是,DNA、RNA、金属离子或代谢产物等小分子也参与了某些蛋白间的相互作用,进行蛋白活性的调节,或直接与蛋白质结合发挥生物学功能,酵母双杂交、TAP 等方法对这类相互作用似乎无能为力,而基于质谱的定量蛋白质组学方法则能继续发挥其自身优势。
2.4 蛋白质芯片 蛋白芯质片技术的基本原理是将各种蛋白质有序地固定于滴定板、滤膜和载玻片等各种载体上成为检测用的芯片,然后,用标记了特定荧光抗菌素的蛋白质或其他成分与芯片作用,漂洗将未能与芯片上的蛋白质互补结合的成分洗去,再利用荧光扫描仪或激光共聚焦扫描技术,测定芯片上各点的荧光强度,通过荧光强度分析蛋白质与蛋白质之间相互作用的关系,并最终达到测定各种蛋白质功能的目的。Zhu 等[23]置备了高密度的酵母蛋白质芯片,涵盖了近5 800 种蛋白,用以研究钙调素结合蛋白。理论上蛋白芯片除了可以研究蛋白间的相互作用外,还可以研究蛋白- 脂类、蛋白- 核酸以及蛋白- 配体间的结合,但目前应用还不够广泛,仅用于CaM、磷脂相互作用蛋白、结构域之间以及抗原与抗体的相互作用研究等[23~26]。随后,Ptacek 等[27]还研究了酵母激酶的相互作用蛋白,发现了近4 000 个磷酸化反应,涉及到1 325 种不同的蛋白 。
2.5 基于生物信息学的分析方法 生物信息学的方法大都基于从已知数据库中分析比较未知蛋白的功能及其相关的相互作用蛋白,已知的数据库包括细菌、酵母的全部基因序列或已知的相互作用蛋白。比较基因簇的同源性推测蛋白功能(conserved gene neighborhood, GN )[28~29],相关基因具有相似的系统发生模式(co-occurrence of genes, CO)[30],相互作用蛋白的基因可能相互融合(gene fusion events, GF)[31~32] 等,以这些为依据采用适合的软件就可对大规模的蛋白质-蛋白质相互作用进行分析,目前已有许多网站开发了这样的功能(表1)。生物信息学分析无需具体的实验操作,往往是在整个基因组规模上进行研究,可获得较大的信息量。另外根据蛋白晶体结构的互补性,也可以推测蛋白间可能的相互作用,但遗憾的是这些方法以基因的同源性为前提,若数据库中没有同源基因则无法比较。目前大规模研究产生的数据都没有达到饱和,整合所有的研究数据而不是分析单一的实验结果,才能得到更加清晰有用的生物学信息。值得一提的是,在细胞中有些蛋白质的相互作用是瞬时的、不稳定的,以实验为基础的研究方法很难捕捉到这种相互作用,而基于生物信息学的分析则能弥补这一不足。
表1 蛋白质相互作用分析相关数据库及网站
网站 资源类型 网址
DIP 蛋白质相互作用http: //dip.doe-mbi.uda.edu
INTERACT 蛋白质相互作用http: //bioinf.man.ac.uk/interactpr.htm
ProNet 蛋白质相互作用http: //pronet.doubletwist.com/
MIPS 蛋白质相互作用http:
//www.mips.biochem.mpg.de/proj/yeast/tables/interactoin/index.htm
Proteome 蛋白质相互作用http: //www.proteome.com
Bind 蛋白质相互作用http: //www.binddb.org
String 基因共定位http: //www.bork.embl-heidelberg.de/string/
CoGs 种系发生谱http: //www.ncbi.nlm.nih.gov/CoG/
2.6 其他方法 具有相关功能或相互作用的蛋白质他们的基因有可能受到了相同的调控,在进化过程中也保持一定的相似性。因此,可以通过检测 mRNA
在特定时期的表达量,分析相关的基因产物是否具有相互作用关系。Stuart 等[33]研究了人、果蝇及线虫的基因共表达模式,发现22 163 种共表达关系并且在进化上它们也表现出一定的保守性。必须指出的是,有些蛋白的表达与mRNA可能不存在相互关系,mRNA 的共表达不反映蛋白间的相互作用;另外所得数据的分析也是一个繁琐的任务。
除了上述大规模分析蛋白质相互作用的方法外,最近还涌现出一些其他的方法。多维蛋白鉴定技术(multidimensional protein identification technology, MudPIT)[34],通过确定蛋白质的亚细胞定位来研究其相互作用等[35]。免疫共沉淀、GST pull-down、 Far-western、化学交联等目前虽不适于大规模研究,但小范围的分析方法可对得到的结果进行补充印证。众多的研究方法可以从不同的角度研究蛋白质组中的蛋白相互作用,弥补单个方法的不足,从而得到尽可能完整的蛋白质相互作用图谱。
3 展望
高等动物基因组研究表明,调节生物体复杂性的不是基因数量的增加,而是基于更为复杂的蛋白质-蛋白质间发生的相互作用。建立起这些分子之间的相互作用及调节网络是后基因组时代的首要任务。这种相互作用的研究包括稳定复合物的亲和力、半衰期及瞬间的相互作用,而且这些复合物中蛋白质的相互作用不是一成不变的,他们存在着动态变化。因此,最终的目标是通过这些作用网络,加上基因动态表达研究、蛋白质表达量分析、蛋白定位以及基因功能分析等,最终了解细胞内各种生理反应的发生及调节机制,揭示生命的本质。目前研究大规模蛋白质相互作用的方法还主要是酵母双杂交及亲和层析,但研究者也发现,所得到的实验结果不能很好的匹配,仅有很少的重叠。这一方面说明蛋白质相互作用的复杂及多样性;另一方面也表明实验方法本身的不确定性及低重复性,对某些相互作用可能还无能为力。因此,对于大规模的蛋白质相互作用研究,不同方法的有机结合将给研究者提供更为丰富、准确的结果。
全文下载:http://www.bbioo.com/bbs/viewthread.php?tid=29316
4.参考文献
[1] Drewes G, Bouwmeester T. Global approaches to protein–protein
interactions. Curr Opin Cell Biol, 2003, 15(2): 199~205
[2] Bauer A, Kuster B. Affinity purification-mass spectrometry.Powerful tools
for the characterization of protein complexes.
[3]Eur J Biochem, 2003, 270(4): 570~578 [3] Fields S, Song O K. A novel
genetic system to detect protein- protein interactions. Nature, 1989,
340(6230): 245~246
[4] Uetz P, Giot L, Cagney G, et al. A comprehensive analysis of
protein-protein interactions in Saccharomyces cerevisiae. Nature, 2000,
403(6770): 623~627
[5] Ito T, Chiba T, Ozawa R, et al. A comprehensive two-hybrid analysis to
explore the yeast protein interactome. Proc Natl Acad Sci USA, 2001, 98(8):
4569~4574
[6] von Mering C, Krause R, Sne B, et al. Comparative assessment of
large-scale data sets of protein–protein interactions. Nature, 2002,
417(6887): 399~403
[7] Walhout A J, Temple G F, Brasch M A, et al. Gateway™ Recombinational
Cloning: Application to the cloning of large numbers of open reading frames or
ORFeomes. Methods Enzymol, 2000, 328: 575~592
[8] Rain J C, Selig L, De Reuse H, et al. The protein-protein interaction map
of Helicobacter pylori. Nature, 2001, 409 (6817): 211~215
[9] Giot L, Bader J S, Brouwer C, et al. A protein interaction map of
Drosophila melanogaster. Science, 2003, 302(5651): 1727~1736
[10] Li S M, Armstrong C M, Bertin N, et al. A map of the interactome network
of the metazoan C. elegans. Science, 2004, 303(5657): 540~543
[11] Stelzl U, Worm U, Lalowski M, et al. A human proteinprotein interaction
network: a resource for annotating the proteome. Cell, 2005, 122(6): 957~968
[12] Rual J F, Venkatesan K, Hao T, et al. Towards a proteomescale map of the
human protein–protein interaction network. Nature, 2005, 437(7062): 1173~1178
[13] Rigaut G, Shevchenko A, Rutz B, et al. A generic protein purification
method for protein complex characterization and proteome exploration. Nat
Biotechnol, 1999, 17(10): 1030~1032
[14] Bauer A, Kuster B. Affinity purification-mass spectrometry. Powerful
tools for the characterization of protein complexes. Eur J Biochem, 2003,
270(4): 570~578
[15] Dziembowski A, Seraphin B. Recent developments in the analysis of protein
complexes. FEBS Lett, 2004, 556(1-3): 1~6
[16] Gavin A C, Bösche M, Krause R, et al. Functional organization of the
yeast proteome by systematic analysis of protein complexes. Nature, 2002,
415(6868): 141~147
[17] Forler D, Kocher T, Rode M, et al. An efficient protein complex
purification method for functional proteomics in higher eukaryotes. Nat
Biotechnol, 2003, 21(1): 89~92
[18] Zhou D, Ren J X, Ryan T M, et al. Rapid tagging of endogenous mouse genes
by recombineering and ES cell complementation of tetraploid blastocysts.
Nucleic Acids Res, 2004, 32(16): e128
[19] Honey S, Schneider B L, Schieltz D M, et al. A novel multiple affinity
purification tag and its use in identification of proteins associated with a
cyclin-CDK complex. Nucleic Acids Res, 2001, 29(4): E24
[20] Ho Y, Gruhler A, Heilbut A, et al. Systematic identification of protein
complexes in Saccharomyces cerevisiae by mass spectrometry. Nature, 2002,
415(6868): 180~183
[21] Zhong J, Haynes P A, Zhang S, et al. Development of a system for the
study of protein-protein interactions in planta: characterization of a
TATA-box binding protein complex in Oryza sativa. J Proteome Res, 2003, 2(5):
514~522 [22] Blagoev B, Ong S E, Kratchmarova I, et al. Temporal analysis of
phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat
Biotechnol, 2004, 22(9): 1139~1145
[23] Zhu H, Bilgin M, Bangham R, et al. Global analysis of protein activities
using proteome chips. Science, 2001, 293 (5537): 2101~2105
[24] Espejo A, Cote J, Bednarek A, et al. A protein-domain microarray
identifies novel protein–protein interactions. Biochem J, 2002, 367(Pt3):
697~702
[25] Hiller R, Laffer S, Harwanegg C, et al. Microarrayed allergen molecules:
diagnostic gatekeepers for allergy treatment. FASEB J, 2002, 16(3): 414~416
[26] Sreekumar A, Nyati M K, Varambally S, et al. Profiling of cancer cells
using protein microarrays: discovery of novel radiation-regulated proteins.
Cancer Res, 2001, 61(20): 7585~7593
[27] Ptacek J, Devgan G, Michaud G, et al. Global analysis of protein
phosphorylation in yeast. Nature, 2005, 438(7068): 679~684
关键词:蛋白质相互作用;酵母双杂交;串联亲和纯化
The advance in research methods for large-scale protein-protein interactions
GUAN Wei, WANG Jian, HE Fu-Chundefined
(Beijing Institute of Radiation Medicine, Beijing 100850, China)
Abstract: In 2000, the first network of large-scale protein-protein
interactions was accomplished in yeast. It was a historic moment and became
another hot spot of protein research. Subsequently, such proteins interaction
maps were also brought to success in the fly, worm and even human during five
years. So rapid progress in large-scale protein-protein interactions turned up
many new angles of view for investigating the mechanisms of cell functions,
actually it also suggested the development and complement of research method
itself. In this article we summarized the progress of techniques in
large-scale protein-protein interactions and showed their differences. Anyway,
the researchers should choose right means to perform their diverse intentions.
Key words: protein-protein interaction; yeast two hybrid; tandem affinity
purification
人类基因组测序的完成标志着一个新的生物学研究时代——后基因组时代(post-genomic era)的来临。科学家们的研究热点又回到蛋白质上,全基因组的序列信息并不足以解释及推测细胞的各种生命现象,蛋白质才是细胞活性及功能的最终执行者。构成细胞的所有组成蛋白有多少;他们的动态相互作用关系又如何;细胞内所有蛋白即蛋白质组又是怎样组织在一起形成有功能、有秩序的网络结构?
这些问题的解决才是最终阐明细胞中各种生命活动机制的关键所在[1]。
1 大规模蛋白质相互作用研究的目的及意义
生物体内各种生命信息由不同的基因经转录、翻译传递到相应的蛋白质上并使其具有各自的生化特性及生物学活性;但每个蛋白质并不是独立的在细胞中完成被赋予的功能,它们在细胞中通常与其他蛋白质相互作用形成大的复合体,在特定的时间和空间内完成特定的功能,而且有些蛋白质的功能只有在复合体形成后才能发挥出来,如依赖于构象变化或翻译后修饰的蛋白质功能[2]
;另一方面,某些蛋白质可能参与了不止一个的复合体,简单的两两相互作用研究就不足以阐明这种更为复杂的相互作用,因此,大规模、高通量的蛋白质相互作用研究应运而生,其目的是在细胞的特定生理条件下,从一个蛋白到多个蛋白,从一个复合体到多个复合体,进而描绘出整个蛋白质组中蛋白间相互作用的网络图。基于这些作用关系,科学家们才能从真正意义上阐明一个蛋白质的功能,才有可能研究细胞中某一生理活动中所有相关蛋白质的变化及作用机制。大规模蛋白质相互作用的研究还有助于了解细胞中不同生命活动之间的相互关系。
2 大规模蛋白质相互作用的研究策略及方法
研究蛋白质相互作用时要根据不同的实验目的及条件选择不同的实施策略。研究已知蛋白间的相互作用人们关注的是蛋白间能否发生结合,实验本身更趋向于验证性,因此,应选择操作性强、可信度高、接近生理条件的技术方法,尽量减少实验本身带来的假阴性或假阳性。若研究者想得到细胞中能与某一蛋白结合的所有未知蛋白质,则实验的关键是得到细胞中含有靶蛋白的复合体,经过有效地分离纯化后,逐一鉴定复合物中的各个组分;或者采用一对多的筛选方法,如酵母双杂交,在建立的蛋白表达文库中检测可能与靶蛋白相互作用的蛋白质。值得注意的是在这一策略中实验结果的进一步验证是必不可少的。目前用于大规模研究蛋白质间相互作用的方法包括酵母双杂交、串联亲和纯化、质谱鉴定、蛋白芯片以及基于生物信息学的分析方法等。
2.1 酵母双杂交 该系统由Fields 和Song[3]首先在研究真核基因转录调控时建立。利用真核生长转录因子的两个不同的结构域:DNA结合结构域(DNAbinding domain)和转录激活结构域(transcription-activating domain),分别与目标蛋白X 及可能与目标蛋白相互作用的蛋白Y 相连,并共同转入酵母细胞。如果蛋白X与Y能够发生相互作用就能使转录因子原来分开的两部分结合,形成完整的活性形式从而激活下游报告基因。通过检测报告基因的表达产物就可判断两种蛋白是否发生相互作用[4~5]。
这一经典的蛋白相互作用研究方法接近于体内环境,那些瞬时、不稳定的两两相互作用也可以被检测到,并且与内源蛋白的表达无关[6]。鉴于这些优点并结合简便高效的Gateway表达载体构建方法[7],在大规模的蛋白质-蛋白质相互作用研究中酵母双杂交系统得到了最为广泛的应用。Rain 等[8]用该法绘制了人类胃肠道病原菌Helicobacter pylori 的大规模蛋白质相互作用图谱。在261 种蛋白中,确立了 1200 种相互作用关系,涵盖了整个蛋白质组得46. 6%。随后,Giot 等[9]和Li 等[10]在果蝇和线虫中也成功地研究了大规模的蛋白质相互作用。除了对模式生物的研究外,Stelzl 等[11]和Raul 等[12]则先后分析了人脑组织、人已知ORF中大规模的蛋白质相互作用网络。但酵母双杂交方法本身也有一定的局限性:(1)不能研究具有自激活特性的蛋白质;(2)只能检测两个蛋白间的相互作用;(3)检测的相互作用需发生在细胞核内,对于不能定位到细胞核中的蛋白质无法研究;(4)大部分实验中有将近50%的假阳性率,且推测的相互作用仅有3% 在两种以上的实验中得到验证。为了弥补方法本身的缺点及局限性,研究者也不断地对其进行完善和改进。Stelzl 等[11]在研究中就采用了以下的策略:(1)选择不同功能、不同大小的蛋白作为诱饵,以确保所选靶蛋白在整个蛋白质组中的代表性;(2)筛选过程中采用两轮杂交的方法:第一轮以混合诱饵(8 个)对文库进行筛选,结果呈阳性的克隆再进行一对一的第二轮杂交,这样既降低了工作量又提高了结果的准确性;(3)pull-down,免疫共沉淀对酵母双杂交的结果进行体内的相互作用验证;(4)利用生物信息学的方法对结果进行系统分析,包括基因的染色体定位、蛋白质作用网络的拓扑结构分析等,从多方面分析结果的可信度。据此,他们最终确认了911 对高可信度的相互作用涉及到401 种蛋白,数据分析中设立了6 个标准来判定得到的结果,大大提高了酵母双杂交实验结果的可信度。
2.2 串联亲和纯化 Rigaut 等[13]首次采用串联亲和纯化技术(tandem affinity
purification,TAP)研究了蛋白间的相互作用,与其他融合标签方法的最大不同是该方法选用了两个连续的标签而不是通常意义上的一个。标签共分三部分:蛋白A 、C B P (calmodulin binding peptide,钙调素结合多肽)和中间连接的TEV 酶识别的酶切位点。带有TAP 标签的融合蛋白在细胞中表达与内源相互作用蛋白形成复合物,细胞裂解后经IgG 偶联的层析柱纯化,蛋白A 与IgG 特异结合,从而使含有标签的复合物得到第一次纯化。为了除去与柱填料非特异结合的蛋白,用TEV 酶切割分离蛋白A 标签,使含有靶蛋白的复合物与层析柱分离并经过钙调素偶联的亲和层析柱进行第二次纯化,靶蛋白上的CBP标签可与钙调素结合;在加入过量螯合剂EGTA后CBP 与亲和层析柱分离使含有靶蛋白的复合体得到分离纯化, 经SDS-PAGE,复合物中的各个蛋白被分开,切胶、胰酶消化后,即可通过质谱鉴定复合物中各个蛋白的氨基酸序列(图1)。该方法的优点:(1)不需要过多的背景知识就可以得到大量含靶蛋白的复合体;(2)蛋白表达及与复合物的结合都接近生理水平,是一种检测体内蛋白相互作用的方法;(3)TAP 采用两步亲和纯化,提高了纯化产物的特异性。一般在2L 的酵母培养基中(细胞湿重约10g)可以分离纯化得到足够一维电泳及质谱分析的蛋白复合物[ 1 5 ] 。Gavin 等 [16]采用TAP 结合质谱鉴定,对啤酒酵母的多蛋白复合物进行了大规模的研究。共分析了 1 739 个基因其中与人类基因相关的有1 143 个,纯化了589 种蛋白,经生物信息学分析,它们分属于 232 个不同的多蛋白复合体;在细胞中具有新功能的为344 个,231 个蛋白的功能以前未见报道。由于TAP/MS方法的高灵敏性使得仅有15个拷贝的蛋白也能被检测到,鉴定的蛋白分子量从6.6kDa 到 559kDa,等电点从3.9 到12.4。这一研究提供了真核生物中二元以上的蛋白相互作用网络,从中得到的有关其功能的各种信息也能为药物的筛选提供依据。TAP 技术的出现有效地促进了酵母中大规模的蛋白鉴定和纯化工作,但是其在高等真核生物中的应用却处于滞后状态,这是因为在真核细胞中表达的内源靶蛋白与带有标签的蛋白会发生竞争作用,影响相互作用的研究。因此,Forler 等[17]将RNAi 与TAP 技术联用在果蝇的S2 细胞(Drosoph ila melanogaster)中研究了蛋白质间的相互作用,该策略避免了内源蛋白的干扰,进而从高等真核细胞中分离并鉴定了相应的蛋白复合体,实验表明,蛋白纯化的特异性及有效性均得到提高,采用这种方法他们还对靶蛋白生物功能进行了分析。iTAP(TAP 与RNAi)保持了TAP 方法在原核生物中应用的一些优势:(1)基于标签的亲和纯化方法;(2)用TAP 标记的蛋白替代内源基因表达产物;但由基因本身的启动子调控蛋白的表达量,这一点在真核细胞中还无法实现。对于这部分相互作用的研究,人们多采用逆转录病毒载体,将带有标签的靶蛋白引入细胞中。由于逆转录病毒载体能够稳定的整合在宿主细胞的基因组中并且拷贝量较低,因此,外源蛋白可以近似于生理水平的低表达,进而避免由于高表达而出现的假阳性结果。此外Zhou等[18]在原代细胞中做了有益的尝试,将TAP标签通过基因敲入技术引入小鼠中,使得该技术能够应用于更多的基因并进行组织或细胞类型特异的复合体研究。
图1 TAP亲和层析纯化原理[14]
注:A:标签中的ProteinA 与固化的IgG 结合缓冲液淋洗去除不能结合的杂蛋白,TEV 酶切分离ProteinA
和靶蛋白;B:利用CBP(Calmodulin binding peptide)-Calmodulin
的相互作用进一步纯化复合物,缓冲液洗去杂蛋白。加入过量的螯合剂(EGTA)螯合Ca2+,使纯化的复合物与层析柱分离。
TAP 纯化时存在蛋白污染问题,很难判断这些蛋白是真正的相互作用,还是蛋白处理过程中的副产品。为此,有人建议用三标签的方法[19],但实验表明该法也不能排除蛋白的污染。其他标签也有应用,比较流行的是Flag[20]。GST、His6、生物素等也分别用于蛋白纯化,但由于他们有限的亲和力及较高的背景,在大规模的蛋白质相互作用研究中未得到广泛应用。选择何种标签也许是个难题,每种标签都有自身的弱点:大标签适于规模较大的相互作用研究,但可能影响蛋白的正常折叠和功能;小标签可能会更大程度的保持蛋白本身的特性,但在表达纯化时通常含量较低[21]
2.3 质谱在蛋白质相互作用分析中的应用 在蛋白质相互作用的研究中,质谱最初只是作为鉴定蛋白质的一种快速、高效的方法:混合的蛋白样品首先经过纯化分离、然后通过SDS-PAGE
将复合体中的组分分开,随后对感兴趣的蛋白进行酶解;得到的肽段在质谱中进行鉴定,从而确定样品中的目标蛋白;但由于分离方法的灵敏度及技术本身的局限,对于那些相互作用较弱、分子量较大或偏碱性的蛋白往往不能达到理想的分析效果,增加了实验结果的假阴性。定量蛋白质组技术的出现解决了这一问题,而该技术的策略正是较好的利用了质谱的高灵敏度。目前已有许多种研究蛋白相互作用的定量蛋白质组学方法,以下就稳定同位素标记方法为例进行说明。稳定的同位素之间具有“轻”、“重” 之分,而其化学性质又保持相同,这就使得有同位素标记的蛋白在分离纯化时具有相同的特性,而在随后的质谱鉴定中又能根据质量数的差异被区分开。通常采用的同位素标记为2H、13C、15N 以及 18O。加入同位素标记的方法主要分为:代谢标记法(stable isotope-labelled animo acids in cell culture, SILAC)和化学标记法(isotope-coded affinity tagging, ICAT)。前者是在培养细胞的过程中加入同位素标记(体内标记) ;后者则是在提取细胞内总蛋白并经过亲和纯化后加入同位素标记(体外标记)。最后两种方法都将得到的对照及实验组样品混合,进行质谱分析。由于同位素“ 轻”、“重” 差异, 质谱能很容易的区分并鉴定实验组中特异的蛋白质。该方法中,亲和纯化过程是最大限度的保留实验组中的差异蛋白而非排除污染蛋白,系统本身的灵敏度在质谱分析中得到保证,这就避免了传统的定性蛋白质组技术中分离方法本身固有的局限性,大大提高了实验结果的灵敏度和准确性。Blagoev等[22]采用SILAC 策略研究了在EGF 刺激细胞后,能够与 Grb2 的SH2结构域结合的相互作用蛋白。两组细胞分别在含有不同同位素(12C-Arg,13C-Arg)的培养基中生长一段时间,EGF 刺激其中一组细胞(实验组)10 分钟,随后两组细胞同时裂解、纯化并进行LC-MS/ MS 分析。最终228 个蛋白得到鉴定,其中28 个蛋白在被刺激的实验组中表达水平明显高于对照组。这些蛋白除了已知的信号分子外,还有一些蛋白参与了信号衰减或具有细胞骨架的功能,此外还鉴定到了未知蛋白质。这表明该方法在研究信号通路中具有较大的潜力,值得一提的是,DNA、RNA、金属离子或代谢产物等小分子也参与了某些蛋白间的相互作用,进行蛋白活性的调节,或直接与蛋白质结合发挥生物学功能,酵母双杂交、TAP 等方法对这类相互作用似乎无能为力,而基于质谱的定量蛋白质组学方法则能继续发挥其自身优势。
2.4 蛋白质芯片 蛋白芯质片技术的基本原理是将各种蛋白质有序地固定于滴定板、滤膜和载玻片等各种载体上成为检测用的芯片,然后,用标记了特定荧光抗菌素的蛋白质或其他成分与芯片作用,漂洗将未能与芯片上的蛋白质互补结合的成分洗去,再利用荧光扫描仪或激光共聚焦扫描技术,测定芯片上各点的荧光强度,通过荧光强度分析蛋白质与蛋白质之间相互作用的关系,并最终达到测定各种蛋白质功能的目的。Zhu 等[23]置备了高密度的酵母蛋白质芯片,涵盖了近5 800 种蛋白,用以研究钙调素结合蛋白。理论上蛋白芯片除了可以研究蛋白间的相互作用外,还可以研究蛋白- 脂类、蛋白- 核酸以及蛋白- 配体间的结合,但目前应用还不够广泛,仅用于CaM、磷脂相互作用蛋白、结构域之间以及抗原与抗体的相互作用研究等[23~26]。随后,Ptacek 等[27]还研究了酵母激酶的相互作用蛋白,发现了近4 000 个磷酸化反应,涉及到1 325 种不同的蛋白 。
2.5 基于生物信息学的分析方法 生物信息学的方法大都基于从已知数据库中分析比较未知蛋白的功能及其相关的相互作用蛋白,已知的数据库包括细菌、酵母的全部基因序列或已知的相互作用蛋白。比较基因簇的同源性推测蛋白功能(conserved gene neighborhood, GN )[28~29],相关基因具有相似的系统发生模式(co-occurrence of genes, CO)[30],相互作用蛋白的基因可能相互融合(gene fusion events, GF)[31~32] 等,以这些为依据采用适合的软件就可对大规模的蛋白质-蛋白质相互作用进行分析,目前已有许多网站开发了这样的功能(表1)。生物信息学分析无需具体的实验操作,往往是在整个基因组规模上进行研究,可获得较大的信息量。另外根据蛋白晶体结构的互补性,也可以推测蛋白间可能的相互作用,但遗憾的是这些方法以基因的同源性为前提,若数据库中没有同源基因则无法比较。目前大规模研究产生的数据都没有达到饱和,整合所有的研究数据而不是分析单一的实验结果,才能得到更加清晰有用的生物学信息。值得一提的是,在细胞中有些蛋白质的相互作用是瞬时的、不稳定的,以实验为基础的研究方法很难捕捉到这种相互作用,而基于生物信息学的分析则能弥补这一不足。
表1 蛋白质相互作用分析相关数据库及网站
网站 资源类型 网址
DIP 蛋白质相互作用http: //dip.doe-mbi.uda.edu
INTERACT 蛋白质相互作用http: //bioinf.man.ac.uk/interactpr.htm
ProNet 蛋白质相互作用http: //pronet.doubletwist.com/
MIPS 蛋白质相互作用http:
//www.mips.biochem.mpg.de/proj/yeast/tables/interactoin/index.htm
Proteome 蛋白质相互作用http: //www.proteome.com
Bind 蛋白质相互作用http: //www.binddb.org
String 基因共定位http: //www.bork.embl-heidelberg.de/string/
CoGs 种系发生谱http: //www.ncbi.nlm.nih.gov/CoG/
2.6 其他方法 具有相关功能或相互作用的蛋白质他们的基因有可能受到了相同的调控,在进化过程中也保持一定的相似性。因此,可以通过检测 mRNA
在特定时期的表达量,分析相关的基因产物是否具有相互作用关系。Stuart 等[33]研究了人、果蝇及线虫的基因共表达模式,发现22 163 种共表达关系并且在进化上它们也表现出一定的保守性。必须指出的是,有些蛋白的表达与mRNA可能不存在相互关系,mRNA 的共表达不反映蛋白间的相互作用;另外所得数据的分析也是一个繁琐的任务。
除了上述大规模分析蛋白质相互作用的方法外,最近还涌现出一些其他的方法。多维蛋白鉴定技术(multidimensional protein identification technology, MudPIT)[34],通过确定蛋白质的亚细胞定位来研究其相互作用等[35]。免疫共沉淀、GST pull-down、 Far-western、化学交联等目前虽不适于大规模研究,但小范围的分析方法可对得到的结果进行补充印证。众多的研究方法可以从不同的角度研究蛋白质组中的蛋白相互作用,弥补单个方法的不足,从而得到尽可能完整的蛋白质相互作用图谱。
3 展望
高等动物基因组研究表明,调节生物体复杂性的不是基因数量的增加,而是基于更为复杂的蛋白质-蛋白质间发生的相互作用。建立起这些分子之间的相互作用及调节网络是后基因组时代的首要任务。这种相互作用的研究包括稳定复合物的亲和力、半衰期及瞬间的相互作用,而且这些复合物中蛋白质的相互作用不是一成不变的,他们存在着动态变化。因此,最终的目标是通过这些作用网络,加上基因动态表达研究、蛋白质表达量分析、蛋白定位以及基因功能分析等,最终了解细胞内各种生理反应的发生及调节机制,揭示生命的本质。目前研究大规模蛋白质相互作用的方法还主要是酵母双杂交及亲和层析,但研究者也发现,所得到的实验结果不能很好的匹配,仅有很少的重叠。这一方面说明蛋白质相互作用的复杂及多样性;另一方面也表明实验方法本身的不确定性及低重复性,对某些相互作用可能还无能为力。因此,对于大规模的蛋白质相互作用研究,不同方法的有机结合将给研究者提供更为丰富、准确的结果。
全文下载:http://www.bbioo.com/bbs/viewthread.php?tid=29316
4.参考文献
[1] Drewes G, Bouwmeester T. Global approaches to protein–protein
interactions. Curr Opin Cell Biol, 2003, 15(2): 199~205
[2] Bauer A, Kuster B. Affinity purification-mass spectrometry.Powerful tools
for the characterization of protein complexes.
[3]Eur J Biochem, 2003, 270(4): 570~578 [3] Fields S, Song O K. A novel
genetic system to detect protein- protein interactions. Nature, 1989,
340(6230): 245~246
[4] Uetz P, Giot L, Cagney G, et al. A comprehensive analysis of
protein-protein interactions in Saccharomyces cerevisiae. Nature, 2000,
403(6770): 623~627
[5] Ito T, Chiba T, Ozawa R, et al. A comprehensive two-hybrid analysis to
explore the yeast protein interactome. Proc Natl Acad Sci USA, 2001, 98(8):
4569~4574
[6] von Mering C, Krause R, Sne B, et al. Comparative assessment of
large-scale data sets of protein–protein interactions. Nature, 2002,
417(6887): 399~403
[7] Walhout A J, Temple G F, Brasch M A, et al. Gateway™ Recombinational
Cloning: Application to the cloning of large numbers of open reading frames or
ORFeomes. Methods Enzymol, 2000, 328: 575~592
[8] Rain J C, Selig L, De Reuse H, et al. The protein-protein interaction map
of Helicobacter pylori. Nature, 2001, 409 (6817): 211~215
[9] Giot L, Bader J S, Brouwer C, et al. A protein interaction map of
Drosophila melanogaster. Science, 2003, 302(5651): 1727~1736
[10] Li S M, Armstrong C M, Bertin N, et al. A map of the interactome network
of the metazoan C. elegans. Science, 2004, 303(5657): 540~543
[11] Stelzl U, Worm U, Lalowski M, et al. A human proteinprotein interaction
network: a resource for annotating the proteome. Cell, 2005, 122(6): 957~968
[12] Rual J F, Venkatesan K, Hao T, et al. Towards a proteomescale map of the
human protein–protein interaction network. Nature, 2005, 437(7062): 1173~1178
[13] Rigaut G, Shevchenko A, Rutz B, et al. A generic protein purification
method for protein complex characterization and proteome exploration. Nat
Biotechnol, 1999, 17(10): 1030~1032
[14] Bauer A, Kuster B. Affinity purification-mass spectrometry. Powerful
tools for the characterization of protein complexes. Eur J Biochem, 2003,
270(4): 570~578
[15] Dziembowski A, Seraphin B. Recent developments in the analysis of protein
complexes. FEBS Lett, 2004, 556(1-3): 1~6
[16] Gavin A C, Bösche M, Krause R, et al. Functional organization of the
yeast proteome by systematic analysis of protein complexes. Nature, 2002,
415(6868): 141~147
[17] Forler D, Kocher T, Rode M, et al. An efficient protein complex
purification method for functional proteomics in higher eukaryotes. Nat
Biotechnol, 2003, 21(1): 89~92
[18] Zhou D, Ren J X, Ryan T M, et al. Rapid tagging of endogenous mouse genes
by recombineering and ES cell complementation of tetraploid blastocysts.
Nucleic Acids Res, 2004, 32(16): e128
[19] Honey S, Schneider B L, Schieltz D M, et al. A novel multiple affinity
purification tag and its use in identification of proteins associated with a
cyclin-CDK complex. Nucleic Acids Res, 2001, 29(4): E24
[20] Ho Y, Gruhler A, Heilbut A, et al. Systematic identification of protein
complexes in Saccharomyces cerevisiae by mass spectrometry. Nature, 2002,
415(6868): 180~183
[21] Zhong J, Haynes P A, Zhang S, et al. Development of a system for the
study of protein-protein interactions in planta: characterization of a
TATA-box binding protein complex in Oryza sativa. J Proteome Res, 2003, 2(5):
514~522 [22] Blagoev B, Ong S E, Kratchmarova I, et al. Temporal analysis of
phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat
Biotechnol, 2004, 22(9): 1139~1145
[23] Zhu H, Bilgin M, Bangham R, et al. Global analysis of protein activities
using proteome chips. Science, 2001, 293 (5537): 2101~2105
[24] Espejo A, Cote J, Bednarek A, et al. A protein-domain microarray
identifies novel protein–protein interactions. Biochem J, 2002, 367(Pt3):
697~702
[25] Hiller R, Laffer S, Harwanegg C, et al. Microarrayed allergen molecules:
diagnostic gatekeepers for allergy treatment. FASEB J, 2002, 16(3): 414~416
[26] Sreekumar A, Nyati M K, Varambally S, et al. Profiling of cancer cells
using protein microarrays: discovery of novel radiation-regulated proteins.
Cancer Res, 2001, 61(20): 7585~7593
[27] Ptacek J, Devgan G, Michaud G, et al. Global analysis of protein
phosphorylation in yeast. Nature, 2005, 438(7068): 679~684
![十二烷基二甲基(3-磺丙基)氢氧化铵内盐 [用于生化研究],14933-08-5,≥98%,阿拉丁](https://img1.dxycdn.com/p/s14/2024/0619/475/6370229598169633081.jpg!wh200)



