阳性克隆筛选方法汇总
江苏吉锐
33626
阳性克隆筛选方法汇总
——基于ZFN、TALEN、CRISPR/Cas9基因敲除方法

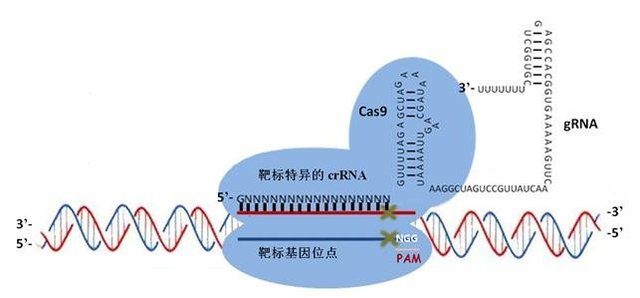
近几年来,基因重组的技术层出不穷,包括TALEN和CRISPR/Cas9在内的重组技术给基础研究提供更为方便和快捷的分子生物学工具。
关于这些技术的具体原理和操作细节,这里不做展开介绍,主要谈一谈在用这些技术进行细胞株构建的过程中,如何做才能加快阳性克隆筛选的步伐,做好这个关键的步。
一,混合克隆pool验证
质粒转染后48-72小时后取转染后细胞的pool 1000个细胞以上离心,去上清,PBS洗一遍,细胞沉淀溶于适量的PBS中,煮5min后模板就制备好了,剩余的pool细胞做有限稀释,一般5块板足够。
分析敲除位点的上下游序列,设计合适的引物PCR扩增目的基因片段,与野生型目的基因杂交后CruiserTM酶切15-20分钟,酶切产物1.5%-2%凝胶电泳。

上图为混合克隆pool酶切检测,由此图可知此混合克隆pool中有敲除成功的细胞,则进行下一步并计算突变率。这一步尤为重要,很多杂志投稿时需要这张图,或者有计算突变率的要求。
二,阳性克隆筛选
上一步采用有限稀释法接种到96孔板中的克隆,待到单个细胞长到几百到1000个细胞左右时,取出其中的一半细胞提取基因组。分析敲除位点的上下游序列,设计合适的引物,并从提取的基因组中扩出目的片段,然后选择CruiserTM或者T7E I进行酶切分析,最后将酶切分析得到的阳性克隆再进行测序进行最终确认。

此图为两个阳性克隆的酶切结果,先酶切再测序不仅可以避免单一方法出现的假阴性,而且可以满足大部分杂志会要求,以免被要求补数据。
为了可以很快的筛选到阳性克隆可以通过如下几个方式去进行优化:
1、尽可能提高单细胞的存活率。
a)对于单个细胞很难存活的细胞,如果是贴壁细胞,可以试着加一些细胞因子促进细胞的分裂;而对于悬浮细胞,则推荐采用Cell Plaza(共培养的原理),都可以极大的提高细胞的存活率;
b)对于很容易老化的细胞,推荐对细胞进行永生化改造,永生化的方法有很多,这里不做具体推荐。
2、采用能够快速抽提微量样品基因组DNA的试剂盒。
这样的试剂盒,市面上也比较多,但是需要注意选择针对样品量比较少的试剂盒,建议选择QIAGEN或者Genloci的微量样品的抽提试剂盒。QIAGEN是老品牌,Genloci这家公司是主要做基因重组的,所以,他们开发的一些产品针对性比较强,而且价格便宜,建议优先使用。
3、设计合理的引物和选用高保真的Taq酶;
引物设计推荐选择靠近靶位点上游100bp和下游200bp处,或者上游200bp和下游100bp处,这样的好处是后面进行酶切分析的时候,对于阳性的克隆,可以很容易观察到2条分开的条带。
Taq酶,建议是用NEB的Fusion或者Q5,毕竟敲除本身很大可能也就是2-3个碱基的缺失,所以,高保证是必要的,防止产生假阳性的结果。
4、选择高特异性的错配酶;
目前用的比较多的是CEL I和T7E I 两种酶,CEL I是植物来源的一种特异性识别碱基错配的酶,活性高,不会产生假阳性。目前市面上只有CruiserTM和Surveyor和两个品牌。其中Surveyor是进口代理产品, 价格较贵,货期较长;CruiserTM价格合适,同时货期短,国内可以2-3天内到货,做到真正质优价廉。
T7E I是NEB的产品,最早并不是为了做基因敲除筛选用的。其最主要的特点是除了识别错配外,还可以识别十字型结构 DNA、Holliday 结构或交叉 DNA、异源双链 DNA,正是因为这个特点,很容易导致假阳性的现象。

图1. Holliday交叉(Holliday junction),无错配结构
5、选择服务好的测序公司
测序公司往往是比较容易忽略的一个环节,很多人送测序后,拿到的测序结果经常发现没信号,就认为是自己的样品有问题,而不会去怀疑测序公司。实际上是错误的思维方式,测序是苦力活,人员流动比较大,所以经常会发现某一段时间的测序结果总是不好,实际上跟样品无关而跟测序公司的人员流动有很大关系。所以,一定要找一些资质比较好的测序公司,同时,发现问题的时,可以考虑换一家公司再测一遍。
测序公司往往是比较容易忽略的一个环节,很多人送测序后,拿到的测序结果经常发现没信号,就认为是自己的样品有问题,而不会去怀疑测序公司。实际上是错误的思维方式,测序是苦力活,人员流动比较大,所以经常会发现某一段时间的测序结果总是不好,实际上跟样品无关而跟测序公司的人员流动有很大关系。所以,一定要找一些资质比较好的测序公司,同时,发现问题的时,可以考虑换一家公司再测一遍。