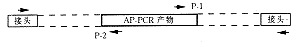基因克隆的常用方法
互联网
基因克隆的常用方法
基因(gene)是遗传物质的最基本单位,也是所有生命活动的基础。不论要揭示某个基因的功能,还是要改变某个基因的功能,都必须首先将所要研究的基因克隆出来。特定基因的克隆是整个基因工程或分子生物学的起点。本文就基因克隆的几种常用方法介绍如下。
1 根据已知序列克隆基因
对已知序列的基因克隆是基因克隆方法中最为简便的一种。获取基因序列多从文献中查取,即将别人报道的基因序列直接作为自己克隆的依据。现在国际上公开发行的杂志一般都不登载整个基因序列,而要求作者在投稿之前将文章中所涉及的基因序列在基因库中注册,拟发表的文章中仅提供该基因在基因库中的注册号(accession number),以便别人参考和查询。目前,世界上主要的基因库有:
(1)EMBL,为设在欧洲分子生物学实验室的基因库.
(2)Genbank ,为设在美国国家卫生研究院(NIH)的基因库.
要克隆某个基因可首先通过Internet查询一下该基因或相关基因是否已经在基因库中注存。查询所有基因文库都是免费的,因而极易将所感兴趣的基因从库中拿出来,根据整个基因序列设计特异的引物,通过 PCR
从基因组中克隆该基因,也可以通过RT- PCR
克隆cDNA。值得注意的是,由于物种和分离株之间的差异,为了保证 PCR
扩增的准确性,有必要采用两步扩增法,即nested PCR
。
根据蛋白质序列也可以将编码该蛋白质的基因扩增出来。在基因文库中注册的蛋白质序列都可以找到相应的DNA或cDNA序列。如蛋白质序列是自己测定的,那么需要设计至少1对简并引物(degenerated primer),从cDNA文库中克隆该基因。以这种方法克隆的基因必须做序列测定才能鉴别所扩增产物的特异性。
另外,在基因克隆之后,如还要进一步做表达研究,所使用的 PCR
酶最好不用Taq DNA聚合酶,而采用其他有自我检测(reading proof)功能的酶,如pfu。这样可以避免由于扩增过程中出现的点突变或终止密码子而导致整个研究结论的错误。
2 根据已知探针克隆基因
这也是基因克隆的一种较直接的方法。首先将探针作放射性或非放射性标记,再将其与用不同内切酶处理的基因组DNA杂交,最后将所识别的片段从胶中切下来,克隆到特定的载体(质粒、噬菌体或病毒)中作序列测定或功能分析。这种方法不但可以将基因克隆出来,还能同时观察该基因在基因组中的拷贝数。但在探针杂交后,要注意高强度(high stringent)漂洗,以避免干扰信号,即保证克隆的特异性,同时节省时间。
3 未知序列的基因打靶
根据已知序列进行基因克隆,多数是重复别人的工作,或者是在别人工作的基础上继续自己的工作,因而不存在新基因的克隆过程。对未知序列的基因克隆才是真正的创造性研究。
3.1 随机引物法克隆未知序列基因
随机引物 PCR (arbitrarily primed PCR ,AP- PCR )首先被用于基因组DNA或RNA的指纹图谱(finger print)分析,后来也有人将这种方法用于克隆与表型相关的基因或mRNA。
该方法的理论依据是:表型受基因支配,在一个生物体发生了表型变化后,其基因组DNA很可能发生变化或出现不同基因的激活或关闭等;另一方面,如在寄生虫的发育过程中,不同发育阶段的虫体所表达的基因很可能不同,如将不同发育阶段的虫体mRNA提取出来,用单一引物(随机引物,其长度不超过16 nt)对不同时期的虫体mRNA进行扩增比较,即可找出导致表型变异的遗传学依据。
这种方法是一种比较 PCR ,它要求至少有2种来自不同表型但又很类似的基因组DNA或mRNA。AP- PCR 扩增后的产物必须99%是一致的,只有个别特异的产物出现在特异的表型个体中。该方法对表型或种源关系相差甚远的生物个体之间没有比较意义。
AP- PCR 的操作一般采用两步法,即前10个循环多以较低的退火温度(annealing temperature)进行扩增,后20个循环则采用较高的退火温度扩增。AP- PCR 产物多较短,一般需高浓度的琼脂糖凝胶检测,也可用聚丙烯酰胺凝胶检测。
如扩增的模板为mRNA,通过比较扩增产物的强度,可以得知该基因在2种生物或同一生物不同发育阶段的表达强度。另外,在AP- PCR 检测到与某一表型相关的基因或基因产物(mRNA)以后,下一步工作就是克隆出整个基因或mRNA。
主要操作程序为:
(1)AP- PCR 产物的提取、克隆及序列测定。首先将AP- PCR 检测到的特定片段从凝胶中切下,提取DNA克隆到适宜的载体内(如TA载体),再测定其核苷酸组成。根据其核苷酸组成设计2个方向相反的引物。引物长度多在20 nt以上;退火温度在60℃以上;
(2)将基因组DNA做适当酶切,然后在其两端连接上相同的接头。根据接头的序列扩增。
3.2 Differential display PCR (DD- PCR )
DD- PCR 是在AP- PCR 基础上发明的一种RT- PCR 方法,主要用于2种或多种类似生物个体在基因表达上的差异分析。其基本原理是:所有真核生物的成熟mRNA都含有不同长度的poly+(A)尾部序列,根据poly+(A)内部的2个核苷酸排列的不同,可以将所有的mRNA分子分为12类。
根据这12种mRNA序列可合成12种相应的反转录引物,即M’N’TTTTTTTT……,用其分别进行反转录,即可将所有mRNA分类合成12种cDNA(于12个试管内),然后再用随机引物,以这12种cDNA分别做模板进行 PCR 扩增,那么与表型相关的mRNA就很容易被发现并克隆出来。
但不论AP- PCR 还是DD- PCR ,都适用于2种种源近似生物或不同发育阶段的同一个体之间的比较。因而,其 PCR 的模板必须是来自2个生物或同一生物的不同发育阶段的mRNA。DD- PCR 的优点是快速、方便,可以检测表达量极低的mRNA,但其技术条件要求较高,所扩增的mRNA的质量不能有差异,即mRNA不应降解。目前这一方法已广泛应用于生物表型相关基因的克隆及比较研究。
3.3 Representative difference analysis PCR (RDA- PCR )
这是一种差减杂交 (subtractive hybridizatio-n)与 PCR 相结合的技术,其整个操作程序完全不同于AP- PCR 和DD- PCR 。前2种方法是将两模板DNA或cDNA分别进行 PCR 扩增,最后通过扩增产物的差异,分离和克隆特异扩增带,其缺点是需要将特异扩增产物从胶中切下来,提取DNA,再扩增后才能克隆。RDA- PCR 只扩增区别于某一表型的特异基因,因而更便于扩增产物的克隆与分析。
其基本原理是:
用一个在种源上相近的基因组将靶基因组中所有共同的基因掩盖起来,而只暴露出特异的基因,在整个反应中只有特异基因能被扩增。
其操作程序为:
(1)用同一限制性内切酶(一般用Bam HI,Bgl II或Hind III)同时处理靶基因组和掩盖基因组DNA,其中掩盖基因组DNA的量至少要2倍于靶基因组DNA的量;
(2)在经酶切的靶基因组片段的两端加上连接头,其序列与酶切产生的粘性末端的序列相对应;
(3)将2个基因组的DNA混合后,高温变性,低温退火。由于掩盖基因组DNA的量远大于靶基因组DNA量,那么与掩盖基因组DNA相同的靶基因就会与掩盖基因形成杂合体,而只有特异的靶基因才能重新组合,不会受掩盖基因的影响;
(4)用Taq DNA聚合酶将粘性末端补齐后,加入与连接子相对应的引物,经过30个左右的 PCR 扩增,就可以将靶基因组中与掩盖基因不同的特异基因扩增出来。这种方法的起始操作程序较复杂,各反应步骤要求准确无误,但其检测的准确度较高。对于基因组内的点突变、重排、插入序列等变化均可检测出来。
4 用特异抗体克隆基因
用抗体克隆基因的关键是抗体的特异性。一般以单克隆抗体最为理想。获取特异抗体的方法主要有:(1)制备识别功能抗原的单克隆抗体;(2)将SDS-PAGE分离的蛋白质特异带切下免疫动物,其抗体只识别该特异蛋白质;(3)用Western-blot法将识别某一蛋白质带的抗体从膜上洗脱下来。在获取理想的抗体后,便可以用这些抗体筛选表达型基因组文库或cDNA文库,从文库中将编码某一特异蛋白质的基因克隆出来。
5 特异基因的功能克隆
它是借助于基因产物即基因所编码的蛋白质的功能将该基因克隆出来的一种方法。基因的功能克隆主要有2种:一是phage display,另一是peptide display。后者的原理与前者基本相同。目前以phage display的应用最为广泛,本文只对这一方法加以介绍。
任何一种病原体,包括细菌、病毒和寄生虫,在其对宿主侵入或致病过程中,病原体本身的蛋白质成分(ligand)需要首先与宿主细胞上的受体(receptor)相互识别连接。
如口蹄疫病毒需要借助于受体细胞上的Heparan sulfate受体,并与其相互作用后才能侵入细胞内;巴贝斯虫裂殖子需借助于宿主的补体作为桥梁,才能与红细胞结合并最后侵入红细胞内。对病原体与宿主受体相互作用成分的特性与功能分析,是制订免疫预防措施及制备阻断药物的前提。phage display法是克隆病原体ligand的一种最为理想的手段。
Phage display的基本操作过程为:
(1)提取某一病原体基因组DNA,用超声波将DNA切割成500~1 500的片段;
(2)将片段末端用T4DNA聚合酶处理后,与载体DNA(phagemid)连接形成噬菌体质粒。用这些质粒与辅助噬菌体(helper phage)同时转染大肠杆菌。phagemid在大肠杆菌内包装成噬菌体,大量繁殖,同时将插入的外源基因片段表达,表达产物主要集中在噬菌体颗粒的表面;
(3)将特定的受体固定于载体上(多为ELISA板),方法同一般的ELISA包被。将噬菌体悬液作适当稀释后,加入平板孔内,感作一段时间,用PBS等缓冲液漂洗。最后,只有表达特异功能蛋白的噬菌体才能与包被在平板上的受体相互结合,那些表达非相关蛋白的噬菌体由于不能与受体连接而被洗脱;
(4)用高强度NaCl液将结合在受体上的噬菌体分离下来,再感染大肠杆菌,便可将编码特异功能蛋白质的基因克隆。
一个真核生物至少含有10万个不同的基因,其中只有10%~15%的基因在不同的发育时期表达,且不同的基因表达的强度相差甚远。
在致病生物中,致病力强的生物,其致病因子(往往是1种或几种功能蛋白或酶)的表达量往往多于致病力弱的生物。从基因组中将人们感兴趣的基因或其转录产物扩增并克隆出来,是进一步对所编码蛋白质作功能分析的关键。本文介绍的几种基因克隆的方法是近几年应用比较广泛的方法。在具体实验中选择哪一种方法,还要根据研究者所要克隆的基因及其所在的基因组和对该基因的研究背景来决定。
(作者:陈启军,解放军农牧大学动物医学系, 长春 130062)