PCR引物设计技巧
互联网
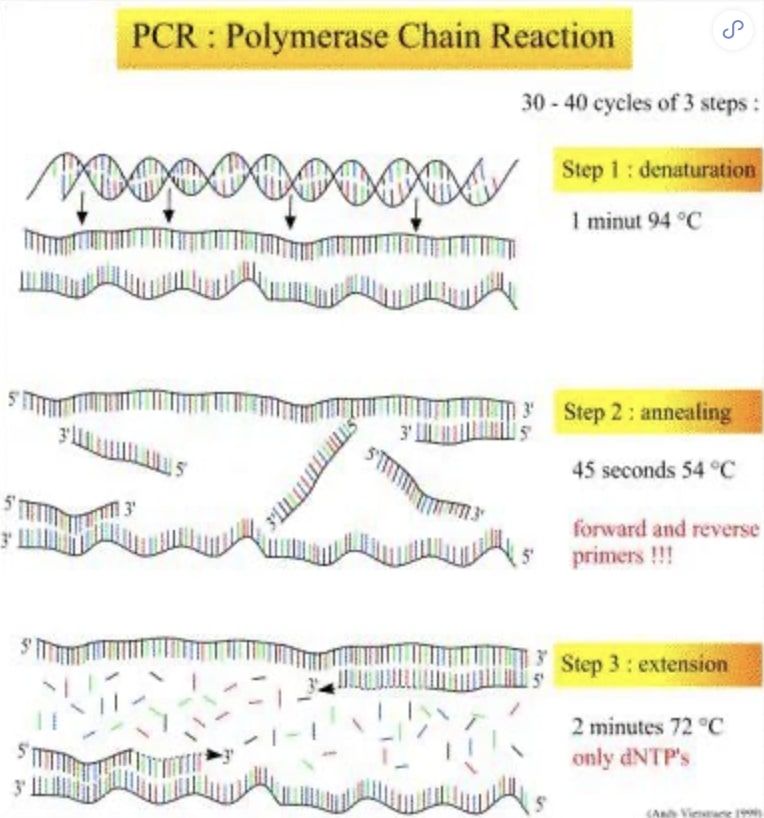
自从1985年美国PE-Cetus公司的人类遗传研究室 Mullis等发明了具有划时代意义的聚合酶链反应(PCP0 以来,PCR已经成为了分子生物学领域最基本也是最重要的技术手段之一[1]。然而能否找到一对合适的核苷酸片段作为引物,使其有效地扩增模板DNA序列,无疑决定着PCR的成败。现在动物遗传育种早已进入了分子时代,在基因水平寻求影响动物遗传表型的新基因突显重要,因此引物设计无疑又成为了寻找新基因的重中之重。
1、引物的设计以及初步筛选
引物的设计与初步筛选基本上通过一些分子生物学软件和相关网站来完成的, 目前运用软件Primer Premier 5 或美国 whitehead 生物医学研究所基因组研究中心在因特网上提供的一款免费在线PCR引物设计程序 Primer 3来设计引物,再用软件Oligo 6进行引物评估,就可以初步获得一组比较满意的引物。但是对于初学者来说,运用软件和程序来设计引物好象无从着手,其实只要我们掌握了引物设计的基本原则和注意事项,所有问题便迎刃而解。
因为无论是软件还是程序,都是以这些基本原则和注意事项为默认标准来进行引物设计的。所以,我们在进行引物设计的时候大可不必在软件和程序的参数上花费过多的时间来思考,如果没有特殊要求我们完全可以把一些参数设为默认值。下面我们主要讨论一下引物设计的原则和注意事项。
①引物的长度一般为15-30 bp,最好在18~24 bp,因为太短易形成错配(False priming) 降低特异性,而太长也会降低特异性,并且降低产量[2]。
②引物在模板内最好具有单一性,也就是说在模板内部没有错配。特别是3‘端,一定要避免连续4个以上的碱基互补错配。
③引物序列的GC 含量最好在40%一60%,且上下游引物序列GC含量的差异不要太大,3’端最后5个碱基最好不要富含GC,特别是连续3个的G或C.
④DNA双链形成所需的自由能AG,应该以5‘端向 3’端递减,3‘端AG最好不要高于9.0 keaf mol[3]。
⑤避免形成稳定的引物二聚体(Dimer and Cross DimeO 和发夹结构(Hairpin),AG高于4.5 keal/mol时易引发 上述两种结构的产生。
⑥ 引物所在的模板区域应该位于外显子区,最好跨越一个内含子区,这样便于对扩增出来的片段进行功能鉴定和表型分析。
⑦ 如果以DNA为模板设计引物,产物长度在100-600 bp比较理想。而以mRNA为模板设计引物时,产物长度在150-300 bp比较理想。
⑧5' 端对PCR影响不太大,可以引进修饰位点和标记物[2]。
只要掌握了以上原则和注意事项,我们可以在软件和程序设计的一组引物中筛选出几对我们需要的目标引物。
2、引物的二次筛选
引物的二次筛选是指在初次筛选出的几对引物中进一步筛选出适合我们进行特异、高效PCR扩增的那对引物。本步应注意以下两点, 一是得到的一系列引物分别在Genebank中进行回检。也就是把每条引物在比对工具(http://www.ncbi.nlm.nih.gov/blast/) 的blastnr中进行同源性检索,弃掉与基因组其它部分同源性比较高的引物,也就是有可能形成错配的引物。一般连续10 bp以上的同源有可能形成比较稳定的错配,特别是引物的3’端应避免连续5-6 bp的同源。二是以mRNA为模板设计引物时要先利用生物信息学的知识大致判断外显子与内含子的剪接位点(例如http://CCR-081.mit.edu/GENESCAN.html的GENESCAN工具或者GeneParser软 ,然后弃掉正好位于剪接位点的引物。
3、引物的最终评估
当我们经过初次筛选和二次筛选后得到的那对引物便可以用于合成,合成后我们经过PCR扩增可以对引物进行最终的评估。一是PCR扩增的特异性和效率。经过PCR条件优化后能否获得特异性条带,即无目的条带之外的多余条带。另外,PCR产物的量是否足够,即无不出带和条带很弱的现象。二是以DNA为模板设计引物时,PCR扩增产物是否与预期PCR产物大小相当。如果相差太大G 于100 b ,有可能是错配产物。三是是否形成引物二聚体带。
我们结合引物最终评估和测序的结果可以对引物设计的成败做出鉴定,为我们以后进行引物设计积累宝贵经验。
4、用比较基因组学分离新基因时引物设计的注意事项
扩增已知基因时经过初次筛选和二次筛选后得到的引物基本上能够满足要求,但是当运用比较基因组学分离新基因时,设计引物还应注意以下两点:① 模板的选择。如果以DNA为模板设计引物, 首先在Genebank中找到与待
根据比较基因组定位的原理,选择研究深入、标记稠密的人和哺乳动物(如小鼠)保守功能基因DNA序列设计引物[51,该引物区段要求在各物种间绝对保守,差异不要大于2 bp,特别是3‘端必须完全同源。如果以电脑克隆策略获得的待分离物种新基因的EST一重叠群为模板设计引物,要求ESTs与信息探针之间同源性大于80%,长度大于100 bp,并且避免在EST的并接部位和可能的外显子与内含子剪接位点处设计引物。②引物序列最好位于相临的外显子区且至少距离外显子与内含子剪接处25 bD以上 ,这样便于对扩增出来的片段进行功能鉴定和表型分析。
[参考文献]
[1]H.A.艾得希主编,田丁等译。PCR技术一DNA扩增的原理与应用【c】。北京:北京医科大学中国协和医科大学联合出版社,1991.
[2]林万明主编。PCR技术操作和应用指南【c】。北京:人民军医出版社,1993.
[3]Oligo软件帮助文件。
[4]Rychlik,W.and Rhoads,R.E.A computer program for choosing optimal oligonucleotides for filter hybridization,sequencing and in vitro amplifi-cation of DNA[J].Nucleic Acids Res,1989,17:8543-8551.
[5]余梅。猪12号染色体上四个新基因的分离、鉴定与物理定位。华中农业大学,2002