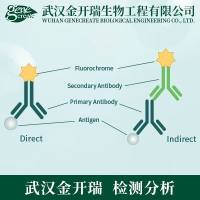
细胞问题解答集锦
互联网
1、舒林酸和尼美舒利的溶解方法的问题:
问:最近在做试验时发现舒林酸和尼美舒利两种药物很难溶解,加入DMSO后可以溶解,但是再加入1640培养液稀释时又出现结晶,而且很难溶解。有什么好的办法吗?
答:(1)可以先把所需药母液加如一个小安剖瓶,再加入培养基,用枪多吹打几分钟就会溶解。
(2)如果还是溶解不了,可以换用乙醇试试。
(3)配制药母液时浓度要尽量高,这样稀释后培养基中的DMSO浓度才越低。
2、细胞因子加入剂量的问题:
问:已经知道某个细胞因子在体内正常的血清浓度为10-12IU/L,临床常用治疗剂量为100IU/kg。现在我们想研究该细胞因子对体外细胞功能的影响。如何决定细胞培养中的该细胞因子加入剂量,谢谢!并请提供原理!
答:临床用药浓度和细胞培养的药物干预浓度无法直接换算,有两方面原因,一是临床用药剂量不等同于血药浓度,即使是已知的血药浓度,也不等同于作用于靶器官/细胞的药物浓度。二是体外细胞学实验是药物作用于孤立的细胞,而细胞在体内要受到众多因素和内环境的影响,对药物的敏感性会有很大的不同。
所以你的问题很难回答,常规的实验思路是这样的:
(1)体外实验,研究药物对细胞是否有作用,如有作用,测出其量效关系和时效关系,进一步研究其作用机理。
(2)体内实验之前,研究一下该药物在体内的药代动力学,毒性等。
(3)动物体内实验,研究药物作用的有效剂量给药方式,进一步阐述其机理。
(4)临床实验。
所以对于你的问题,我的想法是,不必拘泥于同临床用药剂量一致性的问题。以该药物的血清浓度为标尺(10-12IU/L),上下呈10倍药物浓度浮动,如0.001 IU/L、0.01 IU/L、0.1 IU/L、1 IU/L、10 IU/L、100 IU/L做一次六孔板实验,得到大致的有效浓度范围后,按此道理再做一次六孔板实验(倍比关系),就可以确定其作用的量效关系了。最后选定理想的作用剂量研究其对细胞具体功能的影响和机制等。
3、细胞培养中的现象问题:
问:现在做系膜细胞实验,细胞复苏后长势良好,隔一天换液时发现培养瓶底有一层薄膜,晃动培养瓶后,有一张完整的膜脱落,培养液没有变混浊,以为是细胞脱壁呢,去掉膜后观查发现细胞还在,已经快长满一瓶了,然后进行传代,第二天观察发现传的六瓶细胞中有三瓶细胞中又出现了类似的膜。是为什么?
答:我以前也碰过,我认为可能是传的细胞太多,细胞粘连在一起,可能是死亡细胞,以前做流式的时候碰过,也有可能是消化的时候加入胰酶后放的时间太长所致。
4、纯化急性分离的成年大鼠心肌细胞及鉴别方式的问题:
问:我用Langendorff装置通过胶原酶好不容易分离出了成年大鼠心肌细胞,但文献讲这样分的心肌细胞中还有很多杂细胞(成纤维,内皮细胞),我要用这批细胞做流式,需要纯化并鉴别纯度,我查了文章好像用了6%bsa沉降法来纯化的,具体怎样操作呢?
答:我们用胶原酶分离下来细胞后,用200目滤网过滤,待细胞自然沉降,得长杆状细胞就是,好像不用分离纯化。
5、问:线粒体特异性的标记物有哪些?
答:可以用JANUS GREEN B(詹那斯绿)染,线粒体中的细胞色素氧化酶可以氧化其成蓝绿色。这是活体线粒体特异性染料。你的目的是否与此有关?
6、MTT法测细胞黏附率的问题:
问:不少文章提到在他们使用MTT法测细胞黏附率或黏附抑制率时,将细胞种入96孔板孵育前,都预先按分组要求,将细胞与干预药物先一起加入试管中或24孔板中孵育30min,然后再种入96孔板中孵育90min或以上,才测OD值。为什么不一次就加到96孔板中,而是先放在试管或24孔板中孵育一段时间才种96孔板呢?是要起到什么作用呢?
答:预先让药物和细胞孵育,可以让药物充分地作用于细胞,对细胞贴壁功能有足够的影响后再测其粘附抑制率,这样结果比较真实。而直接将药物和细胞一次加到96孔板中,药物作用发挥得不会很充分。
7、线粒体提取问题:
问:我准备做液闪仪测心肌细胞线粒体ANT转运酶活性检测,想从培养所得的细胞中提取线粒体进行检测。看到过一些检测方法,但不知得率是多少,也没有看到出处,有哪位做过,能给个提取线粒体的方案吗?方便的话给个出处。我查到韩国的一篇文献上有方法,但需要超声破膜,我们这没有破膜机器,不方便,所以想找个不用超声破膜的方法。若提得线粒体,如何看它有没有活性呢?用詹那绿B就可以么?另外,因为接着要用液闪仪,可是从细胞里提线粒体是不是会很少啊,液闪仪大概需要多少样本量呢?
答:(1)提取新鲜心肌组织细胞内线粒体的方案:心肌组织切碎后在4℃介质(0.25mol/L蔗糖、10mmol/L Tris-HCl pH7.4、0-4℃)中制备心肌组织匀浆。匀浆经750g、离心10min后留上清,以9000g离心20min后留沉淀,重新悬浮后以9000g再离心20min,弃上清得线粒体.全部操作于4℃进行。
(2)培养细胞提取线粒体。细胞弃去培养液后,用细胞收集液作用2到3分钟,用scraper,将细胞收集起来,6000g,离心15分钟。弃上清,之后加细胞匀浆液,用匀浆器或手动匀浆管匀浆,将装有匀浆液的离心管配平后放入普通离心机,以2500rpm,离心15分钟,缓缓取上清液,移入高速离心管中,保存于有冰块的烧杯中。以17000rpm离心20分钟,弃上清,留取沉淀物。加入0.25mol/L蔗糖与0.003mol/L氯化钙液lml,用吸管吹打成悬液,以17000rpm离心20分钟,将上清吸入另一试管中,留取沉淀物,加入0.1ml 0.25mol/L蔗糖与0.003mol/L氯化钙溶液混匀成悬液,即得线粒体。
(3)液闪仪我没有用过,不过我觉得提取的线粒体数目应该足够你做(每个心肌细胞中都有>1000个的线粒体)。具体的上样标准我觉得需要查你使用的液闪仪的说明书。
8、问:盐酸乙醇该怎么配置呢?
答:0.5%的溶液应为重量百分比,即100g溶液中含有0.5g的盐酸溶液。由于浓盐酸的浓度不为100%,且乙醇溶液的密度不为1,所以楼上的配制方法得出的浓度为盐酸乙醇溶液(1->200),而不是0.5%。
标准的配制方法应为:取一定体积的浓盐酸(约相当于HCl 5g,有浓度、密度,就可以计算出体积了),加乙醇至100g,摇匀,即得。
9、微量全血培养的问题:
问:我现在想利用微量全血培养进行染色体畸变分析,在培养时加入全血0.3ml,但培养过程中感觉有溶血现象,细胞收集后量非常少。可以提供一些经验吗?
答:不知道你用的是什么培养基,我用1640培养,一般不溶血,加某些药物如PHA可促使溶血.实际上溶血对实验结果只有好处没有坏处的,全血可以做几个稀释梯度1:2、1:3、1:5等。细胞收集后量应该与这个也没有关系,一般1ml血大概只有10的6次方个细胞,还有就是细胞收集过程中容易出现问题。
11、langerhans细胞的培养问题:
问:最近准备做免疫耐受方面的实验,需要langerhans细胞培养,一直没发现有这方面资料。请问有细胞系吗?原代培养可行不?
答:据所查资料显示:langerhans细胞好象没有现成的细胞系,一般都是原代分离培养的:
我们实验室的分离方法:
(1)食管粘膜单细胞悬液制备:取距食管癌组织边缘3cm以外的相对正常食管粘膜约3cm,用小镊子及眼科剪钝性分离食管粘膜上皮,并剪成约1cm2的上皮片,室温下0.2g/L的EDTA 孵育10min,37℃,2.5g/L胰蛋白酶孵育30min,用小镊子轻轻将上皮层撕脱,将上皮片加入含100u/ml青霉素100mg/L链霉素,0.8g/LDNase的Hanks平衡盐溶液中,用眼科剪将上皮充分剪碎,用巴斯德吸管充分吹打,通过100 目钢网过滤成单细胞悬液,台盼蓝染色证实活细胞比例大于70%。
(2)不连续密度梯度离心液制备:首先用甲基泛影酸钠和聚蔗糖分别配制高、低不同密度的保存液,高密度液由78.2g/L甲基泛影酸钠与127.1g/L聚蔗糖配制而成,低密度液由44.6g/L甲基泛影酸钠与72. 5g/L聚蔗糖配制而成。然后按高密度液与低密度液的质量比例分别为0 %∶100 %;25 %∶75 %;50 %∶50 %;75 %∶25 %;100 %∶0 % ,配制出密度为1.100、1.089、1.071、1.060、1.052g/cm3的液体。
(3)离心:按密度由低至高的顺序将配制的离心液各1ml分别缓缓加入10ml离心管,保持不同密度的液体不相互混合,不同液面之间有明显分层。将1ml单细胞悬液(细胞数约5x106) 缓缓加入离心液上层,4000r/min离心60min。
(4)LC判定:收集离心所得各层细胞,Hanks液洗2次,涂片,丙酮固定10min。采用鼠抗人CD1a单克隆抗体,按照免疫组化一般方法,进行免疫组化染色。胞膜呈棕黄色的为LC。
再附上langerhans细胞的一些背景资料:
人体langerhans’细胞(LC)来源于骨髓的树突细胞,通过循环迁移到真皮层,然后进入真皮层。LC作为抗原提呈细胞在皮肤局部防御及介导敏感接触反应方面起着重要作用。Langerhan spcell(LCs)主要侵犯淋巴结副皮质区,向滤泡间区、髓质区呈片状或灶性浸润,常见淋巴滤泡残留,有时形成大片坏死,纤维增生似肉芽或结节状,LCs灶有不等量嗜酸性粒细胞浸润。根据LCs形态分以下4种类型:
a.肾形和卵圆形核:细胞较大,胞浆丰富,分界不清,胞浆淡伊红染或透明,核卵圆形或肾形,染色质细,核膜薄而清晰,核仁未见,核分裂象罕见。
b.咖啡豆样核:细胞中等大,胞浆淡伊红染,胞界不清,核呈咖啡豆样有深浅不一的核沟。核膜清晰b.核染色质细,核仁不易见,形似咖啡豆,核分裂象少见。
c.混合细胞型:由咖啡豆样细胞和大圆形细胞组成,约各占1/2,形态同b。
d.大圆形细胞核:细胞大,核大圆形,核膜清晰,核染色质淡而细,核仁明显,常呈嗜酸性,有1-2个小核仁,核分裂象易见,胞浆丰富淡染或空淡,亦有少数咖啡豆样细胞,伴有巨核或多核巨细胞。
12、有效保持细胞的原始体积的问题:
问:我想测定细胞悬液中的细胞体积分布以及数量,由于不能当天就测,只能第二天运到较远的实验室去测,我把细胞泡在含有乙醇、甲醇、甲醛等混合固定剂的细胞保存液中(临床上做液基细胞学用的)。但在测定时发现细胞体积明显比预想的小!不知是不是保存过程中细胞发生了皱缩?有什么好方法可以有效的保持细胞的原始大小(细胞活性无所谓)?
答:以细胞悬浮液:100%ethonal,1:1。最后浓度为50%並存放於4度冰箱中,可放一周,並可測到一些蛋白活性。
13、检菌离心问题:
问:检菌实验要几天才出效果,我们检过1640和FBS均三四天才混浊,难道是双抗的作用?我们昨天做了7721传代,过程中离心出了点问题1000r、10分效果都不是太好,但前天同样的操作1000r5分效果就很好,是不是胰酶消化的影响,因为前天消化时把胰酶吸出了又加了hanks,昨天没吸直接加的hanks离心,今天观察有受菌感染的迹象。
答:你加的Hanks是中和胰酶中含的EDTA的吧,不然也没必要在消化后加入Hanks.你前天是消化时把胰酶吸出了,而下一次没吸出而直接离心,那肯定在离心的过程中胰酶还在继续作用,所以导致你离心时间的延长。
7721这种细胞株消化时间很短,我们实验室用0.25%和0.033%(当然浓度可以更小,文献是0.02%)的EDTA联合消化,加入1mL左右的消化液(铺满瓶底即可),消化时间一般只有30S左右,看到瓶底出现白芒芒的,并开始出现细胞针孔状的间隙,此时就可以把胰酶吸出来,直接滴入几滴血清转动下瓶子终止消化,并随后加入1640吹打,即可全部吹下,我们也没离心除去EDTA,长的也一直很好!离心只是在你加入胰酶后消化时间久了细胞都随胰酶液而漂下,为减少细胞损失才进行操作的,如果你掌握的好细胞是不会在胰酶作用的时候漂下来,所以也就不需要离心,减少了污染的机会!还有放入孵箱的时候要把瓶口拎紧,避免很多人拿进拿出造成污染,虽然说松一点可以利用孵箱的二氧化碳调节PH,但是7721长的很快,两三天即可长满,传代操作也很即时,PH对细胞影响不大!(当然得保证你的培养基PH是正常的,最好两个星期用完)。7721很好养,只要楼主细心操作,肯定实验会顺利开展的。
14、葡聚糖的溶解问题:
问:葡聚糖是用水溶吗?我今天用水溶解葡聚糖,浓度是1mg/ml,四十度水浴,但就是不溶解,瓶底总是有颗粒状的不溶物,不知道是什么原因?或者是我的溶剂用错了?
答:葡聚糖是麦芽中非淀粉质多糖的主要组成部分,其占麦芽干物质的5%~8%,是通过β(1、3)、β(1、4)糖苷键随机排列的线性连接而成的。麦芽中水不溶性的β-葡聚糖主要存在于完整胚乳细胞壁中,热水可溶性β-葡聚糖,主要存在于胚乳细胞之间和蛋白质混合在一起。β-葡聚糖在水中溶解时,浓度低时直接与水分子相互作用增加溶液粘度,浓度大时,β-葡聚糖分子自身相互作用缠绕成网状结构,能吸收水分子形成凝胶,使溶液的粘度大大的增加。
可以用热水试试,不行就用1N氢氧化钠溶解看看;0.1mol/L的就可以;也可以用PBS溶。
15、流式细胞仪用于荧光定量的问题:
问:目前用线粒体标记Mitotracker标记了线粒体,不知道能不能用流式细胞术来定量荧光强度,能够反应线粒体的活性或者数量吗?
答:可以的,我就是用GreenFM标记线粒体数量的,用流式完全可以用平均荧光强度和峰的位置来反应。
16、淋巴细胞分离滋养细胞的问题:
问:我培养的是人绒毛外滋养细胞,用的是胰蛋白酶消化法,用的淋巴细胞分离液来分离,但是离心后,没有看到灰白层。是怎么回事呢?
答:有三个可能的原因:
(1)胰蛋白酶消化后细胞没吹散,最好在消化后的细胞过细胞漏网;
(2)淋巴细胞分离液密度不对 查细胞大小/密度,调整淋巴细胞分离液密度;
(3)溶细胞的溶液可能会对细胞分离产生影响,可用Hank's液。
17、细胞形态的观测方法问题:
问:我在钛板上培养细胞,钛板不透光呀,不能观测细胞在钛板上生长的形态。像MTT感觉只能检测到细胞的数目。我查资料可以荧光染色用荧光显微镜,但是手边没有。用扫描电镜吧又太贵了。
我想细胞能不能一种方法固定住,到别的学校观察到呢,但怎么固定才能保证细胞形态不变,又不掉下来呢,我查看资料要用酒精或戊二醛,却不知道行不行,能不能固定住?如何观测到细胞的形态呢?
答:呵呵,我在人工血管上检测细胞是否黏附的方法可供参考,具体方法如下:
(1)95%酒精固定10分钟;
(2)甲苯胺蓝溶液浸泡30分钟(可惜我不知道浓度,我用的是实验室配好的);
(3)清水漂洗残留的甲苯胺蓝溶液,这时候就可以看到黏附在上面的细胞形态了。如果你还不能确信是否为细胞,可用镊子轻刮表面,刮得下东西的无疑就是细胞了。
18、问:要给大鼠灌维生素a应该用什么溶剂呢?
答:维生素A是脂溶性的物质,易溶于非极性的溶剂如:大豆油,油菜油等植物油。但是制成水溶制剂易于吸收,胆汁酸、胰脂酶、中性脂肪、维生素E及蛋白质均促进维生素A的吸收,拿普通生理盐水、蒸馏水或糖水稀释即可!可以加入一些表面活性剂,制成混悬型的液体制剂,这样即克服了其溶解性,又有利于吸收。
19、G418的配制问题:
问:我买了GIBCO 1g的G418,听说配制时要注意效价。上面标有725μg/mg as is basis。如果要配成100mg/ml的浓度,是不是加入7.25ml的HEPES溶液就行了?
答:标有725μg/mg as is basis的意思是每mg粉末中的有效成分为725μg,所以,如果要配成100mg/ml的浓度,直接加入7.25ml的HEPES溶液就行了,但要注意,HEPES液的pH值,我的经验是pH值在7.5左右最好!
20、细胞爬片后saponin破膜问题:
问:我想在细胞爬片后用0.1%的saponin破膜,但是我不知道破膜时间应该多久,我该怎么控制阿?我养的是肝癌细胞株,请指教。
答:0.1%的saponin破膜,10分钟即可。不知道你之后细胞要进行何种处理,如果还要加入抗体,则10min后吸掉或倒掉皂素时残留一点,因为皂素打孔是瞬时效果,打开后不是一直打开哦。抗体甚至可以用皂素来配置,以保证在皂素存在时(一边打孔一边抗体就进入细胞)。我以前都是这么做的,效果不错。
21、从上海细胞库购买细胞的问题:
问:我3个星期前从上海细胞库订购了一瓶SK-BR-3细胞,但是至今没有到,每次问都说还没有长好,这个细胞那么难长吗,还有大家有买过这种细胞的吗,它寄过来的时候是用什么培养液?1640还是DMEM,用什么样的血清呢?
答:我从中科院上海细胞库订购过细胞株,应该是可信的。
不同的细胞增殖周期不同,一般当客户联系订购并将资费汇过去(一般1200RMB额外加150RMB邮费),传真汇款单,对方收到后开始解冻细胞,传代培养.如细胞生长快,数日张满便用快件的形式邮寄给你.我订购的细胞生长较慢,等了大概10天,才收到细胞.邮寄时间一般为每周三或周四,24-48h可以收到,可以在周末前收到细胞,进行消化传代培养.
不知你是否已将money汇了过去,如果不是,先联系汇money;如果已经汇了,可能是因为该细胞确实生长慢,或对方解冻传代出了问题,需要重新进行一次.关于培养条件,如果是ATCC的细胞株,上海细胞库则完全按照ATCC的标准培养条件选用相应的培养液和血清。参见网址:http://www.atcc.org/common/catalog/numSearch/numResults.cfm?atccNum=HTB-30
22、问:我想做一个测定红细胞脆性的试验,请问哪一种方法好、人的红细胞脆性正常值是多少?
答:红血球脆性试验(RBC Fragility test)是血液实验室用以检查贫血疾病的项目之一,特别是针对遗传性球狀红血球贫血。本次实验在探讨新鲜检体之立刻反应与经37℃孵育24小時后检体反应结果的相关性及经孵育测试的参考值,并对以不同的转速离心做评估。結果发现经37℃孵育24小時的步骤是不可省略的,因只有如此才可筛检出具轻微症状的病人;我们以25位体检病人求得经37℃孵育24小時结果的参考值,分別是完全溶血(最大抵抗)max:0.40%-0.45%,开始溶血(最小抵抗)min:0.55%-0.6。
23、细胞筛的使用问题:
问:细胞筛如何使用?200目与300目区别大吗?大鼠肺组织经细胞筛研磨后得到的是什么细胞?
答:(1)细胞筛使用:洗干净、但不能泡酸,可以直接用锡箔纸包好湿式消毒,使用时不能干磨,比较大的组织边磨边加缓冲液;
(2)200目与300目区别在于孔的大小,一般地,做流式区别不是很大,但300目的使用时注意不能研磨过分,以免结缔组织过多;
(3)大鼠肺组织经细胞筛研磨后得到的是混合的细胞,包括肺实质细胞、基质细胞、红细胞和少量的白细胞、NK细胞以及巨噬细胞。
24、MTT遇到的问题:
问:我做的是悬浮细胞的MTT,因为细胞趋向凋亡,接种密度比较高,有三种,分别是10000个/孔、100000个/孔、500000个/孔,加10微升MTT培养4小时后,用酸化异丙醇溶解结晶,但是测的吸光度都非常低,只有0.005左右,请问是什么原因呢?
答:我也遇到过类似问题,但OD值也没那么低,差不多0.2。开始也以为是MTT的问题,重新配置之后,仍然低,因此改用20%SDS-50%DMF做裂解液,OD值就高了,没有进一步试验是不是别的原因,现在一直用这种裂解液代替酸化异丙醇了。配置方法可参看下面文献:文献中用的是14%SDS,效果差不多。
请点击查看:MTT方法.pdf
25、流式细胞周期的结果分析:
问:我用流式检测细胞周期变化得到如下结果:
组别 凋亡率 细胞周期
G0/G1 S G2/M
对照组 0.04 78.33 11.49 10.18
药物1组 7.68 68.99 15.16 15.91
DDP组 27.87 50.38 49.34 0.28
联合组 35.88 56.92 42.65 0.42
谁能帮我分析一下这个结果,如果G2期出现0,是实验失败么?
答:给药时对照组与处理组细胞的状态有没有检测,周期分布情况如何?如果比较一致的话你的结果可以说明DDP和联合用药组可以引起S期的阻滞,并引起凋亡。G2期低主要是因为细胞被阻滞在S期不能进入G2/M期,是很好的实验结果,并不是实验失败。
26、HEK 293T细胞的有关资料:
问:我马上就得做六组共转染,开始选择的是HEK 293T细胞,但因为一些问题,暂时决定换成HeLa细胞.就有关HEK 293T细胞的几个问题请问大家:
(1)现在我们实验室有一个293细胞系,以前老板用来做过腺病毒包装,请问此细胞,是否可以用来转染。或者,此细胞仅仅可以用做腺病毒包装,不能表达蛋白。
(2)看了丁香园上的很多前辈的有关293细胞的贴子,有些前辈说293细胞系各细胞株都可以转染细胞,请问是不是这样。
(3)如果我们现有的不能转染,请问用过这个细胞系的前辈是否知道这个能表达外源蛋白的293T细胞系如何得到?
答:(1)293T由于表达大T抗原,所以可以增强带有SV40启动子的基因的表达,所以多用来做过表达试验。
(2)293细胞系各细胞株都可以转染细胞,用钙转的方法就可以,转染效率可在50%以上。
(3)The 293T/17 cell line is a derivative of the 293T (293tsA1609neo) cell line. 293T is a highly transfectable derivative of the 293 cell line into which the temperature sensitive gene for SV40 T-antigen was inserted. 293T cells were cloned and the clones tested with the pBND and pZAP vectors to obtain a line capable of producing high titers of infectious retrovirus, 293T/17. These cells constitutively express the simian virus 40 (SV40) large T antigen, and clone 17 was selected specifically for its high transfectability.
27、关于CHO-K1细胞稳定转染和瞬转COS的问题:
问:我最近一直在瞬转COS细胞分泌表达,上清一直没有表达,也浓缩过再做ELISA WB,都没有。时间挺长了,实在等不及了,就想稳定转染cho-k1细胞系,通过MSX来筛选,加压25UM的MSX,结果还不到两个星期,CHO全死了,不知道怎么回事。如果有表达的话,应该是分泌表达的,但是怎么我总是做不出来呢?质粒纯度和浓度都应该没问题,到底哪里我还没有考虑到?
答:提三点建议:
(1)用MSX筛选之前最好做预实验,摸出其最小有效浓度,然后用这个浓度去筛选阳性克隆;
(2)试试间断筛选,就是筛选一周到10天,换压力培养基为完全培养基,细胞状态好转后,再立刻换成压力培养基;
(3)在检测蛋白水平之前,一定先在mRNA水平予以检测,如果检测出目的基因的转录,再行蛋白水平的检测;据我的经验,好像委托公司进行ELISA法检测更为价廉物美。
28、免疫磁珠分离细胞的问题:
问:现在采用间接免疫磁珠分离干细胞的方法:
一直采用的方法是:
10%BSA封闭一小时,
抗体/培养基稀释 4度孵育半小时
二抗磁珠/pbs稀释 4度孵育半不时
刚分离出的细胞状态还可以,但是养几天就不是很好了,考虑是否是因为操作时间太长。最近看了几篇文献,好像他们用磁珠分离都没有使用封闭的过程。而是在抗体的稀释液中加入0.1%血清。请问这样做是否有道理?
答:我们几乎天天做这个,我觉得最好还是封闭好了再加一抗,不过我们封闭时间只有10分钟,没有必要1小时,那个太长了,本来这个过程就很多操作,不能把时间托的太长,细胞毕竟还是要善待的。封闭10分钟的细胞吊出来之后我们养5天,都没有问题,活的很好。
29、问:做MTT时,monodansylcadaverine(MDC)用什么溶剂溶解比较好?
答:无特殊,用PBS缓冲液等即可。
30、文献中实验方法的问题:
问:我最近准备做3H-TdR细胞增殖试验,查了一些国外文献,如下:
1 mCi of [3H] thymidine was added for another 18 hours and proliferation assessed in triplicates. Data are representative of 3 independent experiments.是不是说:每次试验作三个复孔,重复三次试验呀?
答:你的理解应该差不多。前面一句是说做三个复孔。后面一句说,重复做了三次实验,采用的数据是三次当中比较典型的一次。
31、24孔板里293细胞转染问题:
问:我在24孔板里做293细胞的转染,转染试剂是Invitrogen的Lipofectamine2000。奇怪的是,每次加完DNA-Lipofectamine complex,24个孔当中的后面几个孔(比如最后6个孔)里的细胞就明显有一部分死亡,死亡率是10-30%,通常最严重的是最后两个孔。但是前面的孔里的细胞总是没有问题。用不同的DNA做实验,都发生同样的问题,就是总是后面的孔里细胞死亡,而前面的孔里细胞没问题。是怎么回事呢?
答:我也遇到过类似的情况,如果你在加DNA-Lipofectamine complex时孔板里是没有液体的,会在风机下风干的,很快的,这样你在加到后面的孔时,部分细胞已经干了,所以你先加的很好,后面的就有问题。解决办法就是,你不要一起吸净所有孔的液体,6个孔为一个单位,吸弃孔里的液体,在加DNA-Lipofectamine complex,就可以了。
32、成骨细胞消化后不贴壁的问题:
问:我养的是成骨细胞,消化总是存在着问题,以前是用1ml左右胰酶消化一会儿,再弃去大部分留少量继续消化,但是发现总是培养瓶的四周的细胞消化的很快都漂了起来,可是中间的还没太消化好,这个时候一吹打就有好多细胞吹打不下来,24小时后观察,发现有好多悬浮的细胞没有贴壁,我分析可能是吹打的时候细胞受了损伤,还有那些先消化下来的细胞胰酶作用时间太长了,后来我再消化时就增加了胰酶的量基本覆盖平底0.5-0.6ml,这回消化的程度很同步,吹打也比较容易,但是两天后观察,同样也有很多细胞没有贴壁,这是为什么呢?加的胰酶要倒掉吗,我没有倒,直接加了培养液(含10%血清)吹打。
答:先加入少量胰酶0.5-1ml左右使其在所有细胞上流过,倒掉,去除漂浮的死细胞,然后加入0.5ml左右胰酶,倾斜几次培养瓶,使胰酶在细胞上层来回的走几次,使所有的细胞都能在几乎同一时间接触到胰酶。然后消化到手动瓶子就有细胞脱落时加入培养液,吹至均匀分散。
33、培养骨髓基质干细胞的问题:
问:我抽提的兔骨髓细胞(欲分离骨髓基质干细胞)原代培养的时候。细胞总是生长得不理想,连分瓶都无法实现,我用的是DMEM培养基,是微环境模仿的不对还是什么原因呢?
答:兔骨髓MSC的分离:取成年新西兰兔用3%戊巴比妥钠按1mg/kg的剂量施以全身麻醉,另用1%利多卡因于手术部位施以局麻;无菌操作下于右胫骨上端内侧部用锐性穿刺锥于骨皮质上开出一直径约2-3mm的孔,使通达髓腔;用18号硬膜外穿刺针接5ml注射器,内含3000U/ml的肝素0.1ml,沿骨皮质上的孔穿入髓腔,抽出骨髓血约2ml;抽出的骨髓血用平衡液洗涤两次后,离心180xg,5min;弃去上清液,沉淀用Ham f12-10%FBS培养液重新打匀;用灭菌的0.17mol/L饱和NH4Cl溶液按1∶5的体积比加入已重新打匀的悬液中,4℃下保留10min,取出再离心180xg,5min;弃去上清后,以几滴平衡液再悬浮后,用细胞计数板作细胞计数,确认有核骨髓细胞量达106-107后,即可进行原代培养接种用。
34、问:做膀胱癌脱落细胞FISH,怎样处理收集到的尿液标本?
答:我曾做过尿液细胞培养,以收集患者晨起第一次尿,因为经过一夜尿液浓缩,有形成分最多!以无菌中段尿的标准用无菌离心管收集,若不能很快实验,可短暂保存在4度冰箱内,最好不要超过一个小时!收集后快速离心,超净台下培养液重悬尿沉渣,接种培养瓶即可!如果你不需要培养,而仅仅是拿细胞直接做,就直接做细胞涂片了,当然有甩片机更好了!做涂片离心后可用PBS重悬洗两到三遍,再细胞涂片!尿液量大于300ml,請先离心最后剩下50-100ml并加入等量100%酒精,最后浓度为50%放置4度崩相中,可保存一周。
35、细胞培养箱的HEPA filter培养问题:
问:默认的是6个月,如果已经提示要换HEPA filter了,是不是就必须马上换,一个星期后再换会怎么样吗?HEPA filter最长可用多久?一般多久才换新的?
答:如果你的培养箱中的细胞没有发生频繁的无缘无故的大范围的细菌污染事件,那你的HEPA就可以继续用;如果只是发生了小范围的污染事件而用75%的酒精擦拭后就遏制这种现象,你也可以不用换。HEPA其实就是一张折成β折叠后的虑纸放在圆盘状的物品中的虑器。我们实验室的HEPA我曾换过。不过若是你们的money充足,也可以一年一换(EPA百乐好像卖1000块一个)。
36、问:外周血单核、巨噬细胞的分离和培养方法。
答:以前认为红细胞、粒细胞、淋巴细胞、巨噬细胞等都是分化终末细胞。很难在体外培养生长或仅能短期培养而不能传代,但是近来技术的发展,有血细胞培养纯化的报道。血细胞可从外周血中用梯度离心法获得,也可从脾脏中用机械法切碎过筛分离获得(尤其是淋巴细胞)。从腹腔冲洗可获得较多的巨噬细胞。也可从骨髓中取前体血细胞,然后加上促进单核/巨噬细胞分化和生长的细胞因子,从而诱导前体血细胞向所要的细胞方向分化。
37、SP2/0细胞周围有黑色和亮的小点的问题:
问:我昨天收到的SP2/0细胞,刚收到的时候还观察到有贴壁的细胞,将多余的营养液吸出后放入培养箱培养,今天观察细胞都是悬浮的,虽然部分细胞还是比较圆比较亮,但大多为单个分布,而且细胞周围有很多黑色的和亮的小点,黑色的我估计是死亡的细胞碎片,那亮的是什么呢?是不是污染哦?我将三瓶细胞分开操作的,不会都污染了吧?还有请问各位细胞经过长途运输(3天)后一般要几天才能恢复开始生长?
答:sp2/0细胞还是比较好养的,一般两到三天就能恢复活力开始生长了。建议你800转离心后换瓶培养就可以除去那些黑点了。亮点应该不是污染,我原来养时也出现过,没有什么影响,若是看着心烦,就离心换新瓶吧。
38、问:非洲绿猴肾CV-1细胞,字母C、V分别代表什么?
答:cv=curriculum vitae
细胞名称: CV-1(M)
供体:非洲绿猴肾
细胞形态:MV1LU
特性:豹肺上皮细胞
建立者和年代:Fogh和Luncl 1956年
资料来源:http://210.34.120.10/software/07/10/001/01/00001/cells/cell_42.htm
39、问:sp2/0细胞的外观现象问题:
问:细胞是从别人实验室拿过来养的,拿来的时候状态很差,细胞不圆滑透亮边缘有褶皱,传代的时候扔掉大部分细胞,留下一点培养,7天后发现细胞变圆,今天发现刚从培养箱拿出来的时候,镜下观察发现细胞状态很好,但是1分钟后细胞开始变皱,请大家帮忙分析一下是什么原因?是否是因为状态本身不好,温度变化对它影响很大?
答:你把细胞从37度的环境中拿到室温,总要给细胞一个适应环境温度变化的过程吧!你所说的这个现象是正常的。就是人在夏天的时候从海南到哈尔滨也要有个适应的过程嘛!刚从37度拿出来的时候,细胞状态的确很好,你说的在室温下放置1分钟以后细胞就不圆了,其实再过一会,等细胞适应了室温,状态就应该恢复了。假如过一会还是不行,那就是细胞自身的状态问题了。
40、问:s180细胞是贴壁生长的吗?
答:(1)s180细胞是贴壁生长的;
(2)通常情况下S180由昆明小鼠腹水传代。小鼠腹腔接种1.0x10 5 个S180细胞后第10天,无菌条件下抽取腹水,用PBS稀释后计数,腹腔接种传代。
另外,可以从ATCC上可以查到。
41、问:Lewis鼠源性肺腺癌细胞(LLC)的来源是什么?
答:C57小鼠:This line is widely used as a model for metastasis and is useful for studying the mechanisms of cancer chemotherapeutic agents.
Lewis lung carcinoma is a cell line established from the lung of a C57BL mouse bearing a tumor resulting from an implantation of primary Lewis lung carcinoma.
The cells are reported to be highly tumorigenic, but weakly metastatic in mice.
如果还不详细的话请查阅这篇文献:
Establishment of a cloned line of Lewis lung carcinoma cells adapted to cell culture. Cancer Lett. 11: 63-73, 1980. PubMed: 7226139。是LLC起源的文章。
42、测定细胞的蛋白含量问题:
问:利用大鼠胚胎细胞做药物的摄取实验后,需要根据细胞在每个孔内的量的多少来分析其对药物的摄取情况。而蛋白量可以反映细胞量。有什么好的方法来测定细胞的蛋白量?
答:细胞总蛋白质含量测定广泛用于细胞生长实验以及用作表示酶,受体及细胞外代谢产物特异性活性的度量单位,蛋白质含量测定最常用的方法是Lowry's法和考马斯亮蓝测定法,后者比前者更敏感,细胞用量少,50-10000个细胞即可。具体方法请查阅司徒镇强编写的细胞培养P188-189。
43、细胞三维培养的胶原问题:
问:正在做细胞的三维培养,用的是BD公司的I型胶原和matrix gel胶,但怎么不凝啊?
买的I型胶原是已经溶解在醋酸溶液中的,无菌包装,3.45MG/ML,按照说明书的提示加了0.023倍体积的氢氧化钠中和,然后补加培养基至胶原终浓度为2.5MG/ML,和细胞沉淀混匀后,置于做好的模型内, 室温下10分钟,37度培养箱内 三个小时后,再加6ML培养基,但二十四小时了,胶仍不凝,有的已经散开了,请问我哪个地方做错了?是氢氧化钠的体积不对么?胶凝了以后应该是白色不透明? 二十四小时是不是就应该凝了?如果错了,是错在哪里了?
答:没有用过BD公司的胶原,但使用过Sigma的I型胶原(固体),一般是自己配制成液态,然后根据需要按一定比例添加10x浓缩培养基及血清(保证细胞的营养),用1M浓度的NaOH调节pH值约7.2,再和细胞混合均匀后添加到培养器皿中,直接放到37度培养箱,一般也就是30分钟左右即可凝固,等完全凝固后,再在其上添加培养基。胶凝固后,由于其内添加了培养基,所以一般是粉色的。
另外,你在添加NaOH及培养基后,测没测过胶原的pH值?再者会不会在胶原中添加培养基量太多?培养基占的比例大,也会影响到胶原凝固。
44、不同细胞siRNA转染效率的检测问题:
问:(1)最近在做RNAi,用的siRNA,然后Real-Time PCR,发现抑制率仅20-30%左右,与说明书说的70-90%相去甚远,咨询相关人员后,认为可能是转染效率的问题,如果转染效率仅有20-30%,那抑制20-30%也就不少了。因此想请教一下,一般不同细胞通常转染效率是多少?我用的是MT4(悬浮)、CEM(悬浮)、HepG2(贴壁)听说悬浮细胞转染效率要低,原代更低,贴壁好一些,是否如此?
另外就是siRNA的转染如何确定转染效率?有人建议加上荧光,但是已经定了,好几千块钱,再定也不好;或者阴性对照加上荧光,那样的话如何检测?还有建议用个阳性对照,找个别人都做得很好的,抑制率很明确的基因来做个RNAi,再与自己的比较,有好的建议吗?
答:通常情况下悬浮细胞转染效率较低,贴壁细胞的转染效率因细胞不同,有些很高有些很低。HepG2的DNA转染效率大概有50%,siRNA暂时没做过。如果你有GFP质粒和针对GFP的siRNA共转染,用只转染GFP质粒的孔做对照,可以看出转染效率。如果没有针对GFP的siRNA就只能转染GFP质粒,一般情况下转染DNA好的话,转染siRNA也不会差的。个人觉得细胞本身的性质决定了转染效率。
问:(2)针对GFP的siRNA是不是在设计siRNA时就将GFP的表达序列设计进去?
答:不是的。针对GFP的siRNA和我们通常用的化学合成的siRNA一样,只不过是针对GFP基因的。
45、转染RNA i时的问题:
问:我在转染RNA i时不小心加了双倍的RNA(相对说明书),请问会有什么影响啊?因为还有后续实验,所以在这求助,能不能继续做下去呢?另外invitrogen的stealth siRNA在细胞培液中能作用多久呢?可达到一周么?
答:我的实验合成的siRNA的使用说明:siRNA的工作浓度一般是10-100nM,我准备用50nM做转染。相同的孔板中,不同的量的siRNA加的转染试剂的量是相同的,所以我觉得不存在质粒DNA转染时与转染试剂的比例不同影响转染效率的问题。如果你的siRNA能有效干扰你的目的蛋白的表达,加的量越大应该沉默效果越好.只是合成的siRNA比较贵,多加多花费。合成的siRNA体外瞬时转染,干扰效果大致能维持2-5天左右。
46、克隆形成试验的问题:
问:我的试验是关于放疗增敏的,可实验室没人做过这方面的试验,没办法,有个师姐建议我在丁香园向大家求教,我想做克隆形成试验,我看了一些资料,根据放疗剂量不同,每个培养皿接种的细胞数不一样,我觉得用培养皿污染的可能性很大,所以想用6孔板,如果放疗剂量为2Gy 4Gy 6Gy 8Gy 10Gy,那么每孔应该接种多少细胞啊,我看了一篇文章,作者是先将细胞接种到100mm培养皿,24小时后加药,再作用24小时,然后拿去放疗,之后将细胞消化,台盼兰染色,计数活细胞,然后再接种到60mm培养皿,我觉得每消化一次就是对细胞的损伤,可不可以接种前就计数活细胞,放疗后直接放在孵箱培养,这样的误差明显吗?另外,我想问问在此计数活细胞的目的是什么?
答:做克隆形成实验首先需要在平皿或孔板中接种相同数量的处理过的细胞,所以需要计数细胞后再种于平皿或孔板中。如果在放疗之前就接种细胞,则经过放疗后,就不能保证成活的细胞数量是相同的,因而在计算克隆形成率时分母则不一致。而计数活细胞的目的是为了将经过放疗后已经死亡的细胞过滤掉,只计数活细胞,将活细胞种于平皿或孔板中。经过大概两周的时间,观察细胞的克隆形成情况,克隆形成超过50个的计为一个克隆,计数不同处理的细胞可克隆形成个数,从而得到不同放疗剂量对细胞的影响程度。
47、贴壁细胞怎么做transwell小室侵袭实验?
问:我是用7901胃癌,用那种塑料的,就一个大孔的那种transwell。请问怎么做呢?
答:贴壁的胃癌7901细胞作起来更简单啊,只要你的材料齐全质量好就可以。
材料:侵袭小室(TranswellChanber 孔径8um),人工基质胶(Matrigel)
具体方法:设置加药感染7901胃癌细胞实验组及空白细胞对照组,24h后收取细胞进行实验。同时将Matrigel 40μl均匀的铺在TranswellChanber膜上,37℃ 30min成胶。取对照组和实验组细胞各2x105/ml 0.3ml分别接种到成胶的TranswellChanber,并将TranswellChanber置于24孔板内,板内加入0.6ml含10%小牛血清的1640培养液,24h后取出TranswellChanbe;用棉签头擦掉Matrige胶及胶上细胞,PBS液洗3次,瑞氏染色。
结果判定:200倍光镜下随机计数5个视野的穿膜细胞数,取均值。实验重复3次。
48、细胞复苏后形态不均一的问题:
问:复苏了u937后,发现数量很多与正常u937细胞形态相似,但是体积却大了2-3倍的“细胞”,是出了什么问题呢?
答:人白血病细胞u937生长比较慢,血清浓度要求比较高,一般要用含10%-15%胎牛血清的RPMI1640培养液来培养,因此建议冻存时应加大血清的量,复苏时才能得到高存活率的细胞。另外,DMSO也应选择质量好一些的,否则会影响细胞的生存质量.体积却大了2-3倍,可能由于:
(1)细胞由于冻存液效果不佳,影响了复苏后的细胞生长状态;
(2)由于冻存条件不稳定,如低温冰箱或是液氮的温度变化而影响了细胞发生变;
(3)由于冻存时细胞发生交叉污染,致使细胞在冻存复苏后发生突变;
(4)也可能由于细胞刚刚复苏,不很适应,比较脆弱,而造成细胞突变,建议加大血清,继续培养观察一段时间。
49、问:鸡胚内脏培养后会形成什么细胞?
答:如果用的是胰酶消化,还是用做成纤维细胞的培养基,那么多数还是成纤维细胞。因为这样的条件下成纤维细胞具有生长优势。其生长速度快,所以是优势细胞。但是如果你换用其它酶消化,得到的细胞可能是别的细胞,而非CEF。
50、关于hepG2细胞的问题:
问:(1)我现在刚刚开始尝试养HepG2 长得挺快, 贴壁也很好。可就是胞质里貌似有小颗粒,是细胞的显微结构呢 还是细胞状态不好的表现呢?另外,给有经验的师姐看了我的HepG2,师姐称赞背景比较干净。但是形态更像星状细胞不像HepG2。
答:HepG2是肿瘤细胞,属于多核细胞,你看到有很多的小黑色颗粒,在细胞内部的很可能是细胞核,在细胞与细胞间质之间如果静止不动很可能是肿瘤细胞正常的黑色素分泌物,如果动的话很可能是黑胶虫污染,黑胶虫污染短时间不会影响细胞生长状态,但当细胞生长状态略差或是细胞刚刚传代后会影响到细胞的生长,建议再观察一段时间,具体情况再具体分析. 若是背景比较干净那应该不是黑胶虫污染,细胞形态呈星状可能由于细胞营养不良,所以呈触角状生长,建议加大血清用量,用同一种培养基再继续培养观察一段时间。
问:(2)我养的SH-SY5Y是多核细胞吗?它是神经母细胞瘤,好像分化比较高,保留了很多神经元的特性。我看到我的细胞里也有小黑颗粒,静止不动,不是黑交虫,调焦距变亮。我的细胞成锥形或三角形,黑色颗粒在角上,不知道属于金色太阳说的哪一种?还是什么细胞器呢?还是血清不好?或者是其他情况?
答:SH-SY5Y人骨髓神经母细胞瘤也是属于多核细胞。你看到的小黑颗粒有可能是细胞核,另外小黑颗粒还有一种可能就是由于血清的原因,经验认为:国产血清容易发生黑色素的沉积,而且当血清从-20℃中拿出溶解使用时,最好要按照-20℃——-4℃——室温的过程一步一步来,不要急于速溶,否则细胞间质中很容易产生黑色物质,建议用56℃灭活的10%进口胎牛血清,胰酶消化浓度稍低些,继续观察培养,如果小黑颗粒没有影响细胞生长状态也没就什么大碍了!
51、关于明胶缓冲液的问题:
问:今天购买的sigma公司的c5a,上面说稀释时要在缓冲液里加入明胶,想请问下明胶缓冲液有哪些呢,怎么配置啊(明胶好像不溶于水,加热溶解后会不会再析出?),哪种明胶缓冲液可以作用于细胞啊,有没有pbs明胶缓冲液呢?
答:我们用明胶的目的也一样,主要用于处理细胞培养皿底,帮助原代EC细胞贴壁,配置时使用pbs,高压后融解,4度放置后凝成胶状,37度复温后可溶解使用,但时间久后,反复高压回影响使用效果。
52、COC1细胞的问题:
问:我的COC1细胞已经复苏两个星期了,细胞是活的,却不增殖,不分裂,哪位好心人可以指点一下吗?是什么原因?
答:是否细胞密度太低了,细胞是密度依赖型的,在密度太低的情况下增殖会非常缓慢。可尝试增加血清浓度到20%,或者将细胞消化吹打一次加新鲜培养液培养。
53、离心丢失大量细胞的问题:
问:我分离胚胎小鼠的胰腺,然后胶原酶消化成单个的细胞,离心前细胞大约为1x106,可是1500rpm(大约400g),离心10min后,细胞竟然丢失一半。而且离心后细胞容易聚集在一起,不太容易形成单个细胞,是因为部分细胞死亡了吗?我的实验要求大量的细胞,细胞数要准确。提高离心力对细胞不好,降低离心力又损失细胞,请问有没有好办法解决?
答:离心1500rpm(大约400g)?离心力是否偏大?一般离心细胞的离心力要求低于200g。如此大的离心力,丢失细胞的可能性不大,你的细胞离心后容易聚集在一起,也是这个原因。你的细胞离心后丢失一半,是细胞计数了吗?有没台盼蓝染色计数活细胞比例?下次建议将上清也观察一下,看是否有活细胞存在!个人认为当前细胞丢失可能是因为离心力过大导致细胞死亡所致。至于如何在保证足够的细胞数和细胞正常活力之间选择?个人认为还是应该首先保证细胞活力的正常,这就要求离心力不能超过200g。至于如何在满足这个离心条件下更多的沉积细胞,就要再请人不吝赐教了。
问:(2)昨天又试了一次,离心前细胞大约为1.3x106,1100rpm(大约220g),离心10min,因为我需要细胞很大的密度,所以我仅仅加入100μl培养基在离心管的底部。细胞竟然只剩下0.5x106。我的细胞丢失是否因为很多细胞粘在离心管壁上没有沉到底端?
答:你用的是15mL的离心管来离心的细胞吗?只用了100uL的悬液?离心完后需要弃去上清吗?如何弃的上清?如此较大的细胞密度、微量(100μL)的体积在移取和计数时应该误差会很大吧?!可能细胞在转移的过程中粘在移液器或离心管壁上了造成了丢失。1100rpm(大约220g),离心10min应该可以将细胞离心下来的。
问:(3)我以前用的是1.5ml的EP管及离心半径只有6.5cm的小离心机, 肉眼发现很多细胞是粘在管壁上的,而没有沉到管底。后改用15mL的离心管及大离心机,肉眼没发现细胞粘在管壁上。
这次我用15mL的离心管1100rpm(大约220g),离心10min,上清我是用1ml的枪轻轻吸走的,剩下大约100μl来重悬细胞,然后取5μl稀释20倍记数的。
我把上清转移到小培养皿中,在镜下观察有不少细胞, 然后我把上清1500rpm(400g),离心10min。有少量沉淀。再次把上清转移到小培养皿中,在镜下观察还有细胞。不知道是用小离心机还是大离心机好? 也许细胞在转移上清的过程中丢失了。但是不应该丢失这么多呀?
答:有个问题很重要:消化下来细胞后,细胞是否还是在消化液中来离心?消化酶消化下细胞后,移入15ml标准离心管,立刻加入适量PBS或培养液(3-5倍消化酶)稀释,终止消化酶的作用,消化酶长时间的作用会导致细胞脆性增加,抗机械损伤能力下降甚至死亡。如果没有注意这个细节,可能是导致细胞丢失的主要原因。关于离心的问题,个人的经验是,转数在800-1000rpm,时间5min足够。一般不需要超过1000rpm,时间也不需要太久,一是可以减少细胞的机械损伤和在室外的时间,另外,这样的转数和时间可以保证细胞沉淀的前提下,沉淀细胞的紧密度适当,吸除上清后加入适量培养液,稍加吹打即可分散悬浮。最后关于计数的问题,不知是否使用计数板计数。1ml液体里计数100万细胞,计数即是平均100个细胞/大格,误差较大,20-30个细胞/大格较适宜,即20-30万/ml(加PBS稀释成5ml后混匀再计数较好)。至于离心后100μl重悬再计数更不可以,如细胞数不丢失,即是100万/100μl,计数板上是1000个细胞/大格,根本没法数出来!如果一定还要数,建议5ml PBS重悬后计数,再离心后100μl消化液重悬(不需再数),实验用。
54、ho/PI双染检测贴壁细胞的问题:
问:请问用ho/PI双染检测贴壁细胞,直接在孔版中加染色液,请问加染色液之前是否需要吸出培养液呢?
答:需要吸出培养液。用PBS洗2次后再加入染料。Ho染色30-45min,PI染色5-10min左右。PBS洗后可以上机观察。
55、GFP标记的样品的固定与封片问题:
问:我要用激光共聚焦显微镜观察带有GFP标签的酵母细胞,但是不知道用什么方法固定与封片,有资料说可以用低浓度的多聚甲醛,尽可能短时期地固定(对于细胞约5-10分钟)。标本可以放在甘油封片剂中(约含90%的甘油)。想请教各位达人这个方法可行吗,哪位高人能提供具体详细的固定于封片方法可真就是在赶集不过了。
另外,如果要是用酵母细胞作FRAP的话,如何制备活细胞的涂片呢?
答:4%多聚甲醛,20-50%的甘油缓冲液封片就可以了,有条件也可以买专门的荧光封片剂的。
56、H2O2诱导细胞凋亡局部细胞大片漂浮问题:
问:是大片漂浮,感觉是一下不贴壁似的,就跟吹下来似的,但我并没有吹,是在瓶里养的,刚加入H2O2还没有,过大概几小时就大片漂了,是什么原因呢?我知道高浓度会漂,可我用低的浓度0.04mM也漂,是怎么回事呢?
答:细胞漂浮我认为主要有两种原因,一就是你所说的,H2O2浓度太大导致细胞的大量、迅速的死亡;还有一个原因是你的细胞密度太高,这样细胞抓壁不牢,在外界刺激下也很容易成片脱落。第一种情况你可以通过设定一定的H2O2浓度梯度来筛选,第二种情况则适当降低细胞密度,一定不要让细胞长得过满。
57、外周血白细胞的培养问题:
问:想要培养鸡外周血白细胞,采集外周血分离得到白细胞,如何进行培养?培养液用1640可以吗?一般能存活多少天?是不是还要加别的细胞因子?比如IL-2?
答:分离得到白细胞计数后悬浮到1640完全培养基,一般情况下,不加其它物质,细胞可以生长五天,或更长的时间,培养五天以上细胞表面标志会有一定变化。如果要培养更长的时间就需要加IL-2,可以培养一个月。
58、如何使细胞悬浮的问题:
问:我现在做一个实验使一个成纤维细胞和一个卵母细胞粘附在一起,把消化的细胞放在皿里的时候一会细胞就会粘附在皿底部了,粘不了几个成纤维细胞就贴壁了,我想问加什么试剂如何减缓细胞贴壁?或者是怎么样不让细胞很快贴壁?
答:我建议你参考胚胎干细胞制作拟胚体时的方法,为使细胞悬浮,可将消化后的细胞接种到细菌培养皿中,皿底经过处理细胞不会贴壁,千万不要用细胞培养皿!我们实验室都是这样做悬浮培养的,应该没问题的。