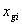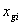基因表达差异的显著性分析
互联网
相关专题
检测基因表达水平
用于检测基因表达水平的 DNA 微阵列实验,应用之一是比较实验,目的是比较两个条件下的基因表达差异,从中识别出与条件相关的特异性基因,例如,识别可用于肿瘤分型的特异基因等。为了提高实验的可靠性,对于同一样本,往往有两次或更多次的重复实验,但是,由于 DNA 微阵列的费用仍然很昂贵,不可能重复足够多的次数来满足实验数据分析的要求,因此需要采用统计方法来分析这些数据。对于这些表达数据的分析,目的就是要识别在两个条件下有显著表达差异的基因。何谓显著表达差异?
通常是指一个基因在两个条件中表达水平的检测值在排除实验、检测等因素外,达到一定的差异,具有统计学意义,同时也具有生物学意义。常用的分析方法有三类,第一类称之为倍数分析,计算每一个基因在两个条件下的 Ratio 值,若大于给定阈值,则为表达差异显著的基因;第二类方法采用统计分析中的 t 检验和方差分析,计算表达差异的置信度,来分析差异是否具有统计显著性;第三类是建模的方法,通过确定两个条件下的模型参数是否相同来判断表达差异的显著性,例如贝叶斯方法。
8.3.1 倍数分析
早期基于 cDNA 微阵列技术的比较实验,用倍数来分析基因表达水平差异,即计算基因在两个条件下表达水平的 Ratio 值。用 表示基因 g 在条件 i 下的表达水平测量值,因此, Ratio 值为 ,可表示基因 g 在条件 1 和 2 下的表达水平差异。对于 cDNA 微阵列实验,是将两个条件下的样本混合后与 cDNA 微阵列进行杂交实验,得到的是成对数据,对每次实验得到的数据计算 ,最后计算重复实验的平均 。
而对于寡核苷酸芯片,首先分别计算两个样本的重复实验的归一化表达水平的平均值,然后计算其 Ratio 值。当 =1 时,基因 g 的表达水平没有改变,而 <1 或 >1 意味着基因 g 在两个条件下存在表达差异,特别是 <1 表示基因在条件 1 是下调的,而 >1 ,表示在条件 1 是上调的。在具体应用中,如果一个基因的平均表达水平在两个条件下的变化超过一个常数,典型的常数是 2 ,即 >2 或 <1/2 ,则认为该基因的表达差异是显著的。
然而,对表达数据仔细考察后可以发现,这样简单的 2 倍法并不能产生最优的结果,因为因子 2 在不同的表达水平上有相当不同的显著性。对于低表达水平的基因,其信噪比太低,用 2 倍法作为判断条件太宽松,而对于高表达基因,条件又太苛刻,往往小于 2 就具有生物学意义。在具体应用中,并没有明确的阈值,往往根据分析的具体要求由数据分析者自行确定。
8.3.2 t 检验
于两个条件下的多次重复实验,为了判断基因的表达差异是否具有显著性,在应用中较多的是采用假设检验,包括两个条件下的 t 检验和多个条件下的方差分析( ANOVA ),这里仅仅介绍 t 检验,关于 ANOVA 请参考相应的统计分析书籍。
零假设为 ,即假设两个条件下的平均表达水平是相等的,与之对应的备选假设是 。
其中 ,为某一条件下的重复实验次数, Xgij 是基因g在第i个条件下第j次重复实验的表达水平测量值。
根据统计量值,可以得到 p 值,它表示在零假设成立的情况下,出现该数据的概率。如果 p 值小于给定的显著性水平,就拒绝零假设,即认为基因 g 在两个条件下的表达差异是显著的。因为在 t 检验中,两个总体平均值之间的距离被样本的标准差归一化,可以克服固定倍数阈值方法的一些缺点。然而,对于 DNA 微阵列数据的 t 检验的基本问题是,即使用当前的高通量检测技术,实验仍然花费很大或者实验过程很冗长,重复次数 经常较小, =2 、 3 的小样本仍然非常普通。由于样本量小,导致总体方差被严重低估,得到的 t 值就较大,因此会导致较高的假发现率 (FDR , False Discovery Rate) ,即通过 t 检验得到的结果中表达差异不显著的基因数目较多。这样,需要更好的分析方法来克服这些缺点。
在 t 假设检验中,经常使用的显著性水平是 p =0.01 ,其意思是在零假设正确的情况下,从总体中进行 100 次抽样,允许有 1 次不满足零假设。对于 DNA 微阵列实验,检测的基因数目巨大,如果微阵列上有 10000 个基因,采用 p =0.01 ,将会有 100 个基因是由于偶然性而被错误认为是有表达差异显著的。这个数目已经可能对后续的生物学分析产生很大的干扰,从而导致 t 检验分析结果的不可靠或失去意义。
为了解决这个问题,可以对 t 检验进行改进,降低由于分母上方差小而带来的错误,因此对 t 检验的计算公式修改如下:
假设 的分布是独立于基因表达水平的。因为较低的表达水平会使 的值较小,导致 值变化较大。为了保证 独立于基因表达水平,在分母上增加 S 0 , 增加 S 0 后可以降低 的方差 。 通过对设计的一组对照样本的分析,可以确定阈值, 大于阈值的基因被认为是表达差异显著的。
8.3.3 贝叶斯分析
由于 DNA 微阵列数据噪声大、波动大,而且在大量数据的背后还有很多相关变量不能被观察到,因此,贝叶斯方法可以用来分析微阵列表达数据。
其中, P(M|D) 表示由观测数据集 D 得到参数化模型 为真的概率,称为后验概率; P(M) 称为先验概率,表示在没有得到任何数据之前所估计的模型 M 为真的概率; P(D|M) 是指似然度,表示从模型 M 得到一个观测数据集 D 的概率。贝叶斯推断是通过参数估计和模型选择来实现任务的,最常用的方法是最大后验概率 (MAP) 估计和最大似然 (ML) 估计。
在用贝叶斯方法分析表达数据时,首先假设在给定条件下,一个基因的表达水平测量值是独立的,并满足正态分布。根据经验,这一假设是合理的,特别是表达水平的对数大致服从对数正态分布。对于重复实验,也可以引入伽玛分布、高斯 / 伽玛混合分布等。一个基因在一种条件下的表达测量值可以用一个正态分布 来建模。对于每个基因在每一种条件下,都对应有一个双参数模型 ,似然函数可以由下式给出:
i 取遍所有的重复测量,重复测量次数为 n ,C表示归一化常数。似然度取决于充分统计量 n 、 和 分别表示重复次数、 n 次重复实验的平均值和方差。
先验概率分布 的选择有几种,一般采用共扼先验分布。先验分布的四个超参数构成向量 ,则
超参数 和 可以分别解释为 的位置和数值范围,V 0 和 分别解释为 的自由度和数值范围。对于 DNA 微阵列数据,采用一个 和 相互不独立的先验分布很有意义。经过一些代数运算,可以推导出后验分布具有与先验分布相同的函数形式:
后验分布的参数以一种合理的方式将先验分布的信息和数据信息结合了起来。后验分布是贝叶斯分析的基本对象,它包含了 和 所有可能取值的相关信息,可以通过多种方法估计 和 。