基因表达数据的获取
互联网
随着 cDNA 微阵列和寡核苷酸芯片(下文没有特别说明时,统称为 DNA 微阵列)等高通量检测技术的发展,我们可以从全基因组水平定量或定性检测基因转录产物 mRNA 。在本章中,基因表达数据特指基于 DNA 微阵列实验得到的反映 mRNA 丰度的数据,而不包括基因表达最终产物――蛋白质丰度的数据。
由于生物体中的细胞种类繁多,同时基因表达具有时空特异性,因此,基因表达数据与基因组数据相比,要更为复杂,数据量更大,数据的增长速度更快。基因表达数据中蕴含着基因活动的信息,可以反映细胞当前的生理状态,例如细胞是处于正常还是恶化状态、药物对肿瘤细胞是否有效等。
对基因表达数据的分析可以获取基因功能和基因表达调控信息,这是生物信息学的重大挑战之一,也是 DNA 微阵列能够在生物医学领域中广泛应用的关键原因之一。
基因表达数据分析的对象是在不同条件下,全部或部分基因的表达数据所构成的数据矩阵。通过对该数据矩阵的分析,可以回答一些生物学问题,例如,基因的功能是什么?在不同条件或不同细胞类型中,哪些基因的表达存在差异?
在特定的条件下,哪些基因的表达发生了显著改变,这些基因受到哪些基因的调节,或者控制哪些基因的表达?哪些基因的表达是细胞状态特异性的,根据它们的行为可以判断细胞的状态(生存、增殖、分化、凋亡、癌变或应激等)等等。
对这些问题的回答,结合其它生物学知识和数据有助于阐明基因的表达调控路径和调控网络。 揭示基因调控路径和网络是生物学和生物信息学共同关注的目标,是系统生物学 (Systems Biology) 研究的核心内容。
目前,对基因表达数据的分析主要是在三个层次上进行:
1 、分析单个基因的表达水平,根据在不同实验条件下,基因表达水平的变化,来判断它的功能,例如,可以根据表达差异的显著性来确定肿瘤分型相关的特异基因。采用的分析方法有统计学中的假设检验等。
2 、考虑基因组合,将基因分组,研究基因的共同功能、相互作用以及协同调控等。多采用聚类分析等方法。
3 、尝试推断潜在的基因调控网络,从机理上解释观察到的基因表达数据。多采用反向工程的方法。
本章首先介绍基因表达数据的来源和预处理方法;然后介绍基因表达数据分析的主要方法,包括表达差异分析、聚类分析和分类等;最后简单介绍从基因表达数据出发研究基因调控网络的一些经典模型。
8 .1 基因表达数据的获取
基因表达数据反映的是直接或间接测量得到的基因转录产物 mRNA 在细胞中的丰度,这些数据可以用于分析哪些基因的表达发生了改变,基因之间有何相关性,在不同条件下基因的活动是如何受影响的。
它们在医学临床诊断、药物疗效判断、揭示疾病发生机制等方面有重要的应用。检测细胞中 mRNA 丰度的方法有 cDNA 微阵列、寡核苷酸芯片、基因表达系列分析( Serial analysis of gene expression , SAGE )、 RT-PCR 等。
目前,高通量检测基因组 mRNA 丰度的方法主要是 cDNA 微阵列、寡核苷酸芯片,它们的原理是相同的,即利用 4 种核苷酸之间两两配对互补的特性,使两条在序列上互补的单核苷酸链形成双链,这个过程被称为杂交。
基本技术路线是:制备芯片,在一个约 1cm 2 大小的玻璃片上,将称为探针的 cDNA 或寡核苷酸片段固定在上面;从细胞或组织中提取 mRNA ,通过 RT-PCR 合成荧光标记的 cDNA ,与芯片杂交;用激光显微镜或荧光显微镜检测杂交后的芯片,获取荧光强度,分析并得到细胞中 mRNA 丰度的信息。
8.1.1 cDNA 微阵列
cDNA 微阵列是在 1995 年由斯坦福大学率先研制成功并应用于基因表达分析的。首先将细胞内的 mRNA 逆转录成 cDNA 并分离,然后将分离得到的所有或部分 cDNA (其长度通常大于 200bp )作为探针,用机器手按照阵列的形式点到玻璃片上。
玻璃片上的每一个点只包含一种 cDNA 分子,这样就制成了 cDNA 微阵列。固定在玻片上的 cDNA 探针可以通过测序得到序列或者其来源是已知的。
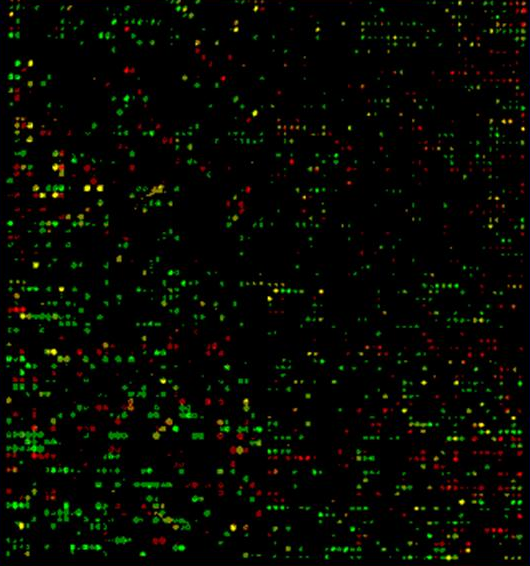
在使用 cDNA 微阵列时,首先提取组织或细胞系中的 mRNA 样本,逆转录成 cDNA 并用荧光素标记;然后把标记混合物加到 cDNA 微阵列上,与探针杂交,杂交过程完成后,清洗微阵列;最后用激光扫描仪扫描并获取荧光图像,对图像进行分析,得到 cDNA 芯片上每一个点的荧光强度值。荧光强度值定量反映了样本中存在的与探针互补的 mRNA 丰度,也就是反映了探针所对应基因的表达水平。
在制造 cDNA 微阵列时,点样点的大小是不能保证完全一样的,点的排列也可能是不规则的,这意味着要比较不同微阵列图像的荧光绝对强度是不合理的,因此通常使用双色荧光系统来纠正点之间的差异。
在制备样本时,使用两个样本,一个称为控制样本( control sample )或对照样本 (reference sample) ,通常用绿色荧光素( Cy3 )标记其 cDNA ,另一个为测量样本,用红色荧光素( Cy5 )标记其 cDNA 。
这两个样本按照相同的实验方案分别制备不同荧光素标记的 cDNA ,并按 1 : 1 的比例混合,然后与 cDNA 微阵列杂交,用不同波长的激光扫描杂交后微阵列,分别获取荧光强度,并成像。
来自两个样本的基因如果以相同水平表达则显示黄色,而如果表达水平有差异,则图像显示红色或绿色。因此, cDNA 微阵列的实验数据反映了两个样本中基因的相对表达水平。由于 Cy3 和 Cy5 的标记效率不相等,以及存在系统噪声等原因,通常需要对 cDNA 微阵列实验中获取的原始图像数据进行归一化。
例如,用 Cy3 、 Cy5 两种荧光素分别标记的一些基因的表达水平相等,那么这些点的实验结果 Cy5/Cy3 荧光强度比率值(以下称 Ratio 值)的期望值为 1 ,但由于得到的 Ratio 值往往不等于 1 ,这些实验偏差可以通过归一化来得到纠正。
对微阵列进行归一化的指导思想包括基于全局强度值调整、强度相关归一化、玻片之间的对比归一化等,归一化方法包括总密度(假设两个样本中的总 RNA 是相等的)、线性回归、 Ratio 统计、迭代 log(ratio) 平均值中心化等,与微阵列扫描系统配套的软件可以完成归一化工作。
cDNA 微阵列实验得到的值反映了基因的相对表达水平,即测量样本与对照样本之间荧光信号强度的比率或者对数化的比率,这是一个无量纲的值,可用于比较一组实验中的基因相对表达水平。
如果对照样本的信号非常低,那么这个比率就可能很大,可能主要是噪声信号,因此它很可能是无意义的。这些数据往往看作是不确定的或异常点,在后续分析时要注意这些数据,根据需要确定是否保留以及如何纠正其值。
8.1.2 寡核苷酸芯片
又称为基因芯片、 DNA 芯片。它是在玻璃片上按阵列固定寡核苷酸探针,这些探针是在片原位合成的。现有产品中应用最广泛的是 Affymetrix 公司制造的 GENECHIP®芯片,它使用一种光掩模技术和传统的 DNA 合成化学的组合以非常高的密度制造寡核苷酸阵列。
例如, Affymetrix 公司的 Human Genome U133 芯片包含了 100 万个不同的寡核苷酸探针,代表了 33000 个人类基因。寡核苷酸芯片主要用于 DNA 多态性检测和基因表达分析,还可以用于微生物基因组的再测序。
寡核苷酸探针的长度通常为 20-25bp ,在检测 mRNA 丰度时可能存在寡核苷酸之间的非特异性交叉杂交,这可能会掩盖杂交信号;此外,对于特定的寡核苷酸,信号强度对于寡核苷酸的碱基组成比较敏感的。
对于第一个问题, Affymetrix 公司的解决办法是采用匹配 / 失配( PM/MM )探针对的方法,即在设计一个特异的寡核苷酸 ( 匹配 ) 时,同时设计一个非特异的寡核苷酸探针,该探针仅仅在中间位置有一个碱基替换(失配),这样可以用 PM 与 MM 之间的差值作为信号强度。为了解决第二个问题,在设计探针时,对于每一个待检测的 mRNA 包含多个寡核苷酸探针,例如设计 11-20 对探针来检测一个转录本。
与 cDNA 微阵列不同的是,杂交实验中与寡核苷酸芯片杂交的是单个样本,而不是 cDNA 微阵列实验中测量样本与对照样本的混合物。寡核苷酸芯片的检测结果有两种,一种用 P/A/M ( Present/Absent/Don't Know )表示,表示有 / 无 / 不确定,另一种用荧光信号强度值表示。
P/A/M 可以用来判断样本中有无特定基因的表达,这个结果对于部分实验,特别是一些定性实验是有意义的,例如判断肿瘤与正常细胞的基因表达差异。当需要对几个不同条件下的基因表达情况进行分析时,对基因表达的相对变化更感兴趣,所以多采用荧光强度值。
有时实验结果中有负值,这是由于前景信号小于背景信号或者背景 / 阴性控制样本的定义不正确造成的, Affymetrix 公司的芯片分析系统会将负值修改成某一固定值。
在分析多个实验条件下的基因表达数据时,与 cDNA 微阵列数据一样,也是一系列测量样本与对照样本之间的信号强度比率或比率的对数值。实验得到的信号强度也是经过归一化的数值,归一化的方法很多,而且一般都包含在芯片扫描系统的图像处理软件中。
cDNA 微阵列或基因芯片在用于基因表达分析时的一个最大优点是高通量性,在一次芯片实验中可以对成千上万个基因的表达进行并行测量。由于实验环节较多,虽然在设计芯片时可以通过添加阴性和阳性探针等手段来评价数据的质量,但是需要提醒的是,数据的可靠性仍然是对数据进行后续分析时必须考虑的一个问题。
8.1.3基因表达数据的网络资源
大量基于 DNA 微阵列实验的基因表达数据是公开发布在 Internet 网上的,尤其是学术机构在发表论文时所用的实验数据都可以免费提供给全世界的研究人员下载使用。
作为学术论文的补充资料在网上发布的数据主要是文本文件或 Excel 格式的文件,这些数据往往都是经过归一化处理后的 Ratio 值或 log 2 (Ratio) ,对于寡核苷酸芯片数据有的是 P/A/M 表示,有的是荧光强度值。
因为这些数据文件没有包含原始的实验方案、实验材料、原始扫描图像、图像处理方法和数据归一化方法等信息,对于要比较或整合分析来自不同研究小组的基因表达数据是非常困难的。
主要原因是 DNA 微阵列并不是在任何客观的个体上测量基因表达水平,大多数测量值仅仅是基因表达的相对变化,而且使用的并不是一个标准化的对照样本。
同时,基因表达数据比基因组序列数据要复杂的多,这些数据仅仅在有具体的关于实验条件的描述时才是有意义的,对于不同的细胞类型,在不同的条件下都有一套转录本。
因此,基于 DNA 微阵列的基因表达数据存储量是非常大的,对于具有 20000 个探针的微阵列实验,以 10um 的分辨率扫描,产生 3 千万个离散的数据点,如果以 tiff 文件贮存,将占用约 60Mb 的硬盘空间。
一方面由于基因表达数据量非常庞大,而且数据中蕴含着丰富的生物学知识,另一方面由于这些数据没有注释,迫切需要一种标准来描述和存贮 DNA 微阵列基因表达数据,同时建立公共的 DNA 微阵列数据仓库。
欧洲生物信息学研究所( EBI )与德国肿瘤研究中心 (DKFZ) 在 1999 年成立了 MGED 讨论组 (The Microarray Gene Expression Data) 。 MGED 是一个国际性的成员联盟,参与人员包括生物学家、计算机科学家、数据分析学家。它的目标是促进由功能基因组学和蛋白组学研究产生的微阵列数据的共享。
当前集中于建立微阵列数据注释和交换的标准,推动微阵列数据库建设和相关软件来实现这些标准,促进高质量的、经过注释的基因表达数据在生命科学领域的共享。该组织开发的微阵列数据标准称为 MIAME(the minimum information about a microarray experiment) ,是对于解释和验证结果所必需的微阵列实验的最小信息描述。 MIAME 不是微阵列实验必须遵循的教条,而是一组指导方针,它将帮助微阵列数据库和数据分析工具的开发。
同时, MGED 组织开发了微阵列基因表达标记语言( MAGE-ML , Microarray Gene Expression - Markup Language ),它是一种语言,用来描述跟基于实验的微阵列信息的通讯。 MAGE-ML 基于 XML ,可以描述微阵列设计、制造、实验组织和实施信息、基因表达数据等。
MIMAE 标准和 MAGE-ML 语言受到了从事 DNA 微阵列开发和应用研究的科研人员和组织的广泛关注。美国 NCBI 的 Gene Expression Omnibus (GEO) 、英国的 EBI 的 ArrayExpress 数据库都采用了该标准,斯坦福微阵列数据库( Stanford Microarray Database , SMD )也正在兼容该标准。
目前,收集、存贮微阵列基因表达数据的最有影响的数据库和网站是 GEO 、 ArrayExpress 和 SMD 。
GEO 是由 NCBI 在 2000 年开发的一个基因表达和杂交微阵列数据仓库,同时作为获取来自不同生物体的基因表达数据的在线资源。到 2004 年 3 月,数据仓库中包含内容有 605 个 Platform , 14391 个 Sample , 816 个 Serial 。
Platform 是关于物理反应物的信息,Sample是关于待检测的样本信息和使用单个Platform产生的数据。Series 是关于样本集的信息,反映样本间的相关性和组织。
ArrayExpress是基于基因表达数据的微阵列公共知识库,目的是存储被注释的数据,当前包含多个基因表达数据集和与实验相关的原始图像集。 ArrayExpress 数据库接受 MAGE-ML 格式的数据递交或者通过 MIAMExpress 的基于 Web 界面注释和递交的数据。
ArrayExpress 提供一个简单的基于 Web 的数据查询界面,并直接与Expession Profiler 数据分析工具相连,可以进行表达数据聚类,和其它类型的 Web 数据挖掘,并将进一步开发多个实验和数据库间的交叉查询。 ArrayExpress 数据库中的数据将与所有由 EBI 维护的或在线的数据库相联接。
SMD 是一个使用 Oracle 作为数据库管理软件的关系数据库。 SMD 存储微阵列实验的原始数据、归一化数据和对应的图像文件。
自从 2002 年 1 月 1 日起,到 2004 年 4 月已包括 85 篇学术论文,超过 3500 个双色点样 cDNA 微阵列的实验数据,并且每年增加 1000 个微阵列实验的数据。另外, SMD 提供数据获取、分析和可视化的界面,目前包括层次聚类和自组织映射等方法,还将加入 k- 平均聚类、单值分解和丢失值归纳等方法。
除了以上 3 个综合性的基因表达数据仓库外,还有一些专门的基因表达数据库,例如 YMD、 ArrayDB 、 BodyMap 、 ExpressDB、 HuGE Index 等,这些数据库收集的数据往往具有物种特异性,使用比较方便。