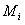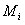基因表达数据的分类分析
互联网
在生物医学中,基于表达数据的肿瘤分型诊断是重要的应用领域。 1999 年, Golub 等人的研究证明了肿瘤亚型之间在基因表达上的差异,通过对一组特异基因的表达检测,可以进行临床诊断,并指导治疗方案的制定。
随后,很多研究组开展了用 DNA 微阵列检测基因表达用于肿瘤诊断的研究,所涉及的肿瘤包括白血病、乳腺癌、肺癌、结肠癌等,有些工作已应用于临床实践。在这些研究中,根据表达数据对样本进行肿瘤亚型分类成为生物信息学的一项重要研究内容。
分类是从训练数据中找出一组能够描述数据集合典型特征的模型(或函数),以便能够分类识别未知数据的归属或类别,即将未知事例映射到某种离散类别之一。
对基于表达数据的肿瘤分类,就是根据已知肿瘤类型的样本数据来构建分类器,然后利用它对新的表达数据进行分类分析,确定肿瘤的类型。基于基因表达数据的肿瘤分型诊断的数据分析主要包括以下过程:
①肿瘤分型特异基因的选择。在 DNA 微阵列实验中,基因数目成千上万,但实际上影响样本分类的,往往只是很少一部分的关键基因,其它的基因往往是不相关的,是冗余的或显著性较小。过多的基因会导致噪声的增加,影响到分类效果。
因此,需要选择对肿瘤分型有效的一组基因,这组基因的表达行为对于诊断肿瘤是特异性的,因此,这组基因也称为信息基因 (informative genes) 。
要确定信息基因并非是件容易的事,常用的方法是前面所述的显著性分析,从不同肿瘤类型的样本中分析基因表达差异的显著性,挑选出显著性高的基因作为信息基因。
但由于基因调控的复杂性和实验控制等因素的影响,这些基因并不能很好地实现分类的目的。此外还有一些方法,例如,采用信息增益来评价基因在分类中的显著性,或采用遗传算法和分类相结合的方法来选择信息基因。
②构建分类器,利用已知类别的数据训练来构建决策规则。方法很多,下面将对常用的方法进行具体介绍。
③检验分类预测的有效性。在对肿瘤分型表达谱数据进行分类分析时,可以通过敏感性和特异性来分析分类结果的好坏。如果用 TP 表示阳性率, FP 表示假阳性率, TN 表示阴性率, FN 表示假阴性率。则敏感性的定义为 TP/TP+FN ,而特异性定义为 TP/TP+FP 。
作为好的分类方法,要求有高的敏感性和特异性,但往往是一对矛盾;此外,对于肿瘤分类问题,还要考虑由于错分带来的风险,需要寻求某种折衷。在对肿瘤样本分类结果进行评价时,通常采用的方法是留一法和独立检验法。
留一法是指每次去除 1 个样本,构建新的分类器,然后对所去除的样本进行分类,统计错误分类的次数。独立检验法将样本分为测试集和训练集。随机将数据分为 k 个子集合,依次取出一个子集作为测试集,而其余的 k-1 个子集合作为训练集,利用分类器,对测试集的样本分别进行分类,计算错误分类的次数,此过程循环 k 次。
1.朴素贝叶斯分类法
这是用于分类的基本方法之一,其基本思想是假设已知各类别的先验概率和似然概率,根据贝叶斯理论,可以计算该样本属于各类别的后验概率,具有最大后验概率的类别就是该样本所属的类别。朴素贝叶斯分类法的关键是要确定先验概率和似然概率,这可以从训练样本中获取。
2.k- 近邻法
于两个条件下的多次重复实验,为了判断基因的表达差异是否具有显著性,在应用中较多的是采用假设检验,包括两个条件下的 t 检验和多个条件下的方差分析( ANOVA ),这里仅仅介绍 t 检验,关于 ANOVA 请参考相应的统计分析书籍。
3.其它分类法
分类方法是机器学习中的常规方法,有很多比较成熟的算法。决策树是归纳学习法中的一种,可以根据训练样本中的属性和值归纳得到布尔型的决策函数,并可以直观地用树的形式表示。
对于多种分类器得到的分类结果,可以采用投票分类法,整合多种分类结果给出最终分类。人工神经网络,特别是 BP 网络是在分类问题中应用最多的一种方法。
<center> <p> </p></center>