双向凝胶电泳
互联网
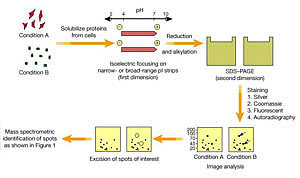
双向凝胶电泳的原理是第一向基于蛋白质的等电点不同用等电聚焦分离,第二向则按分子量的不同用SDS-PAGE分离,把复杂蛋白混合物中的蛋白质在二维平面上分开。近年来经过多方面改进已成为研究蛋白质组的最有使用价值的核心方法。
分离蛋白质组所有蛋白的两个关键参数是其分辨率和可重复性。在目前情况下,双向凝胶电泳的一块胶板(16cm×20cm)可分出3~4千个,甚至1万个可检测的蛋白斑点,这与10万个基因可表达的蛋白数目相比还是太少了。
80年代开始采用固定化pH梯度胶,克服了载体两性电解质阴极漂移等许多缺点而得以建立非常稳定的可以随意精确设定的pH梯度。由于可以建立很窄的pH范围(如0.05U/cm),对特别感兴趣的区域可在较窄的pH范围内做第二轮分析,从而大大提高了分辨率。
此种胶条已有商品生产,因此基本上解决了双向凝胶电泳重复性的问题。这是双向凝胶电泳技术上的一个非常重要的突破。第二向SDS-PAGE有垂直板电泳和水平超薄胶电泳两种做法,可分离10~100kD分子量的蛋白质。
其中灵敏度较高的银染色法可检测到4ng蛋白,最灵敏的还是用同位素标记,20ppm的标记蛋白就可通过其荧光或磷光的强度而测定。
用图像扫描仪、莱赛密度仪、电荷组合装置可把用上述方法得到的蛋白图谱数字化,再经过计算机处理,去除纵向和横向的曳尾以及背景底色,就可以给出所有蛋白斑点的准确位置和强度,得到布满蛋白斑点的图像,即所谓“参考胶图谱”。
蛋白质组研究的主要困难是对用双向凝胶电泳分离出来的蛋白,进行定性和定量的分析。最常用的方法是先把胶上的蛋白印迹到PVDF(polyvinylidene difluoride)膜上后再进行分析,确定它们是已知还是未知蛋白。
现在的分级分析法是先做快速的氨基酸组成分析,也可先做4~5个循环的N末端微量测序,再做氨基酸组成分析;结合在电泳胶板上估计的等点电和分子量,查对数据库中已知蛋白的数据,即可作出判断。
有文献报道,N末端4个残基序列的数据就可以给出很多的信息而得到相当准确的结果。如再结合C末端序列,判断结果的准确性会更高。对分离得到的蛋白质作进一步的确切鉴定需要有足够数量的纯蛋白,电泳时蛋白质已经过了高度纯化。
现在一块胶板可允许上到高达mg数量级的样品,因此每个分离的蛋白斑点可有μg数量的蛋白,这样使本来是微量的蛋白也可希冀被鉴定。
蛋白质的翻译后修饰和加工,是指在肽链合成完成后进行的化学反应,如磷酸化、羟基化、糖基化、二硫键形成,以及最近发现的蛋白质自剪接等等,可能有一百种以上。翻译后修饰和加工对蛋白质的正常生理功能是必需的,它们的变化往往和疾病的发生有关。
用双向凝胶电泳可以进行翻译后修饰的研究,如用32P标记可以研究磷酸化蛋白的变化。双向凝胶电泳中常可发现的蛋白质拖曳现象,很可能是一个蛋白的不同翻译后修饰产物所造成的。拖曳图像变化在疾病诊断上可能提供重要的信息。
双向凝胶电泳技术当前面临的挑战是:
(1)低拷贝蛋白的鉴定。人体的微量蛋白往往还是重要的调节蛋白。除增加双向凝胶电泳灵敏度的方法外,最有希望的还是把介质辅助的激光解吸/离子化质谱用到PVDF膜上,但当前的技术还不足以检出拷贝数低于1000的蛋白质。
(2)极酸或极碱蛋白的分离。
(3)极大(>200kD)或极小(<10kD)蛋白的分离。
(4)难溶蛋白的检测,这类蛋白中包括一些重要的膜蛋白。
(5)得到高质量的双向凝胶电泳需要精湛的技术,因此迫切需要自动二维电泳仪的出现。