


师姐呕心沥血整理的 qRT-PCR 注意事项
生物学霸
大家都在说 qRT-PCR 实验原理、引物设计、结果解读等,而我觉得我应该和大家分享一下 qRT-PCR 的实验操作事项。虽小,但是关乎结果。
在做 qRT-PCR 之前,我们需要对自己的 RNA 以及操作方法有清楚的了解,毕竟我们的努力是希望得到结果的,而不是简单的练练手。所以在做 qRT-PCR 之前,我们需要确定以下问题(部分内容仅适用于 SYBR)。
你确定你的 RNA 没有降解吗?
NanoDrop 2000 只能检测 RNA 的浓度以及纯度,而对于 RNA 的完整性是没法检测的。
RNA(RNA Intesity Number)值可以反映 RNA 的完整性,通过 Agilent 2100 Bioanalyzer system 来检测
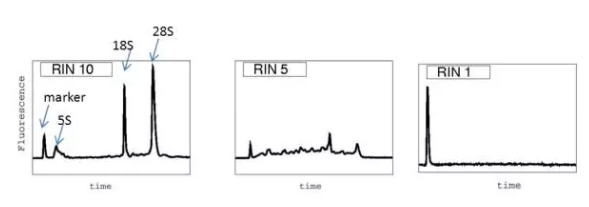
图. 不同 RNA 样本的 RIN 值示意图(真核生物)
但是实验室一般不会有 Agilent 2100 Bioanalyzer,在这种情况下,我们可以通过甲醛胶检测,但是对 RNA 总量的要求高,那么最快速的方法就是用普通的凝胶电泳。要求在 nuclease-free 的环境下,所以需要将电泳槽、溶胶瓶、凝胶托槽以及梳子用 DEPC 水冲洗干净。琼脂糖也是 nuclease-free(只要是新开封的就行),Loading Buffer 尽可能是新开封的,配置 1.2% 的凝胶。
注意,凝胶一定要保证溶解完全,要不然会导致条带不均一。电压太高或者跑太长时间会产热,导致 RNA 降解,所以要合理控制电压和时间。此外,跑胶也可以进一步确定样品中是否有 DNA 的残留,观测点胶孔是否有大量滞留的条带。
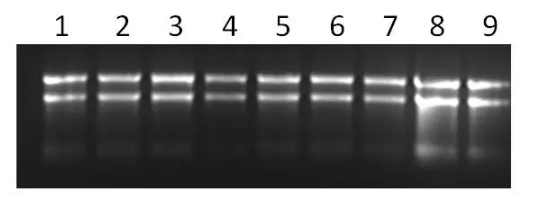
图. 凝胶电泳检测 RNA
你确定你的 cDNA 的浓度吗?
实验室大师兄的经验是每次反转得到的 20 ul 体系的 cDNA 直接稀释 20X,而博后师姐是按照 10X 稀释,我一般是看情况而定。因为每个人提的 RNA 质量不同,反转的水平也分高低,再说反转的技术也未必稳定。
所以在每次拿到反转的 cDNA 后,我首先会稀释 3 倍左右,然后利用管家基因做一次 RT-PCR,循环数一般为 25 cycle,鉴定一下具体浓度,再决定最后的稀释倍数。
你确定你的引物好用吗?
可以通过 qRT-PCR 的溶解曲线,但是呢,这个还是要耗钱的。对于没有很多钱的实验室来说,在拿到很多引物的时候,可以通过普通的 RT-PCR 看看是否是单一条带,鉴定一下引物的特异性。如果实验室不差钱,则可以通过溶解曲线对所有的引物的特异性做一次鉴定。
你确定你的 PCR 板和仪器配套吗?
如果你用的是 BIO-RAD 公司的定量 PCR 仪器,那么你一定要买配套该仪器的 qRT-PCR 板。因为不兼容的 PCR 板会导致结果偏差。因为我的师姐就真的碰到过这种情况。
你确定你的实验条件合适吗?
SYBR 要避免强光照射,所以在加 SYBR 试剂的时候尽量关掉头顶的照明灯,只需借助微光完成就可以。
SYBR 放置 4 ℃ 保存,使用时轻轻上下颠倒混匀即可,避免泡沫产生,切忌用力涡旋。
有些小师妹怕自己加样搞混,喜欢在 PCR 板上划出标志,这样做是不对的。因为你的标记极有可能影响到荧光信号的采集,所以一般我会建议师妹采用实验记录本协助记忆,如下所示。

图.qRT-PCR 加样图
你确定你的操作正确吗?
一定要戴手套,戴手套,戴手套,重要的事情说三遍。
为了减少 SYBR 在光下的曝光,个人喜欢先加入模板, 如下图。根据经验,少量模板的加入,容易造成加样误差。所以为了尽量减少加入少量模板造成的误差,我一般会将样本再稀释一倍,加样的时候多加一倍,减少 H2O2 的加入量。
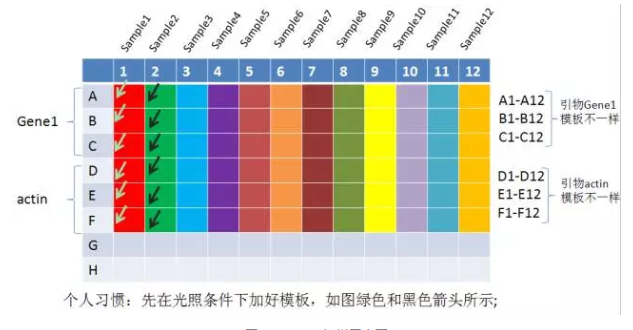
图.qRT-PCR 加样示意图
然后配置 qRT-PCR 体系,如下。
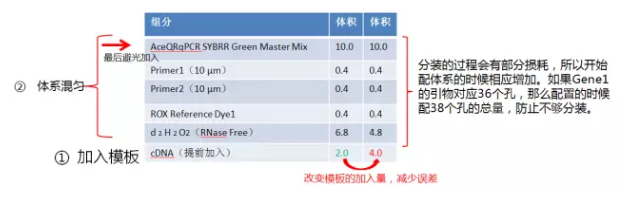
图.qRT-PCR 体系配制图
注:配置过程需在冰上完成。
加好样品后,贴好透明封板膜。尽量不要用手碰透明封板膜的表面,从膜两边空余处操作就行。因为手印也有可能可能影响到荧光信号的采集。然后就是用离心机低速迅速离心 10 S,防止样本挂壁。
好了,基本的操作部分以及注意事项就这么多,希望能帮到大家
<link />






