当 BioEdit 遇上 Excel ,手把手教你批量处理序列!
丁香园
由核苷酸 ATCG 及氨基酸组成的序列代表着核酸和蛋白质最基本的一级结构,对它们的编辑分析是实验室必备的技能。
除了使用让人头大的编程语言及命令行,今天推荐的两款纯交互图形界面软件 BioEdit + Excel 组合效果也毫不逊色,特别是序列的批量处理方面,而且操作更加简便直观,堪称实验室序列编辑分析出奇制胜的双剑客。
本文就组合使用 BioEdit + Excel 批量更改 Fasta 格式文件中的序列名、本地 BLAST、从泛基因组中提取特定基因序列、批量 G+C 含量计算等作具体实例,带大家领略这两款软件组合应用的魔力。
批量更改 Fasta 格式文件中的序列名
许多下游生信分析软件对输入文件中的序列名有严格要求,但是我们所获得数据的序列名常常过于繁琐或者杂乱无章,如从 NCBI 数据库下载的来源于某一物种基因组的基因集。
这里我演示了结合 BioEdit 与 Excel 将某酸球菌属 Acidisphaera 某菌株 As1 的基因集文件(Fasta 格式)中的序列名称按顺序批量改成 As1_1、As1_2、As1_3……形式的快捷方法。
1. Fasta 格式文件的第一行是由大于号「>」打头的文字说明,用于序列标记,从第二行开始为序列本身。
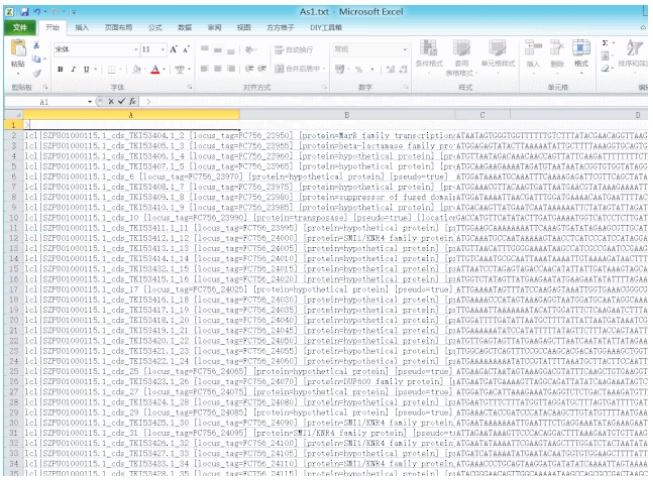
首先用 BioEdit 打开原基因集文件,依次点击 File -> Export -> tab-delimited text (title,tab,sequence),导出为文本,并用 Excel 打开。可以看到原来以「>」打头序列名间隔的序列变成了序列名(A 列)-基因序列(B 列)的行对应形式。

BioEdit 打开并导出 Fasta 格式原基因集文件为制表符(Tab)分隔形式文本
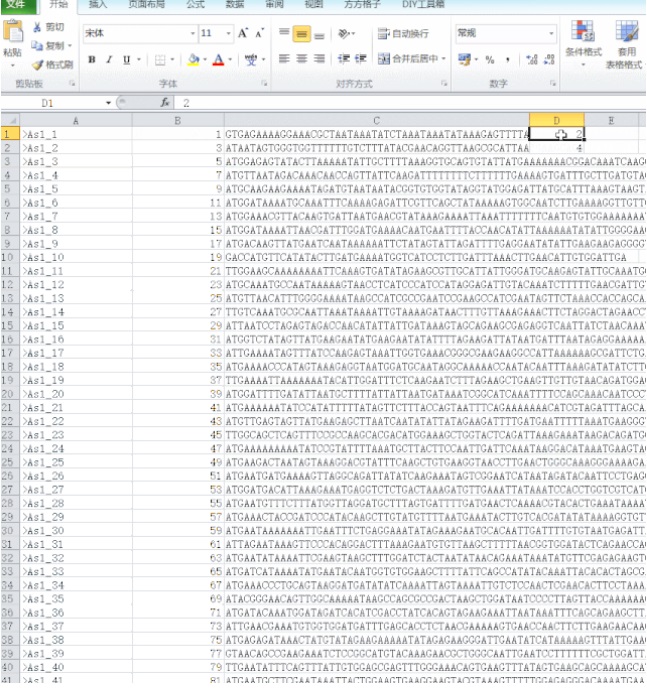
2. 接着,在原 A、B 列之间插入辅助列,并将第一个序列名称(及 A1 单元格)改为「>As1_1」,使用 Excel 的智能填充功能按顺序批量更改原序列名(把鼠标放到单元格右下角,双击符号+),其中「>」是之后重新转换成 Fasta 格式所需要的。
使用 Excel 的智能填充功能按顺序批量更改原序列名
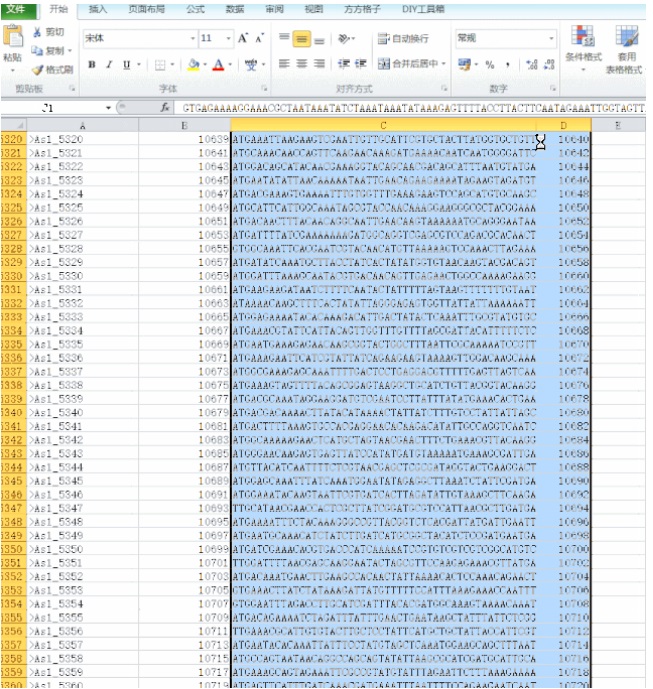
3. 然后,在辅助列 B 列与 D 列分别智能填充升序的奇数和偶数,并将 C、D 列的数据一起移动到 A、B 列下方。
辅助列 B 列与 D 列分别智能填充升序的奇数和偶数
将 C、D 列的数据一起移动到 A、B 列下方
4. 最后,选中辅助列 B 列,依次点击 Excel 的「数据」选项卡 -> 升序 –> 扩展选定区域 –> 排序。
删除辅助列 B 列,这样基因集文件(Fasta 格式)中的序列名就按顺序批量改成 As1_1、As1_2、As1_3……的形式,形式更加简洁容易索引,更重要的是可以直接作为下游全基因组范围分析软件如进化压力分析软件 PosiGene 的输入文件。

利用辅助列重新排序
本地 Blast 及从泛基因组中提取特定基因序列
这个实例中,要从酸球菌属 Acidisphaera 数个菌株(编号为 As1 到 As5)的基因组中批量比对并提取出异源物质代谢中的编码 3 -oxoadipyl-CoA 硫解酶的基因 pcaF,并进行 G+C 含量计算、批量翻译、蛋白质性质预测、多序列联配以及进化压力分析。
1. 首先构建 Acidisphaera spp. 的泛基因组,即将需要分析的菌株所有基因整合到一个 Fasta 文件里。
用 BioEdit 打开 As1 菌株的基因文件 As1.fasta 并选择导入菌株 As2、As3、As4、As5 的基因文件:File -> Import -> Sequence alignment file –> Save as,这样 5 个基因组的基因序列就整合在了一个文件里。
构建泛基因组
2. 接着要收集种子序列(Query sequence),即已知的 PcaF 蛋白序列(可以到 NCBI 里直接搜索),保存到一个文本里。然后在 bioedit 里把基因组种子序列建成本地数据库:Accessory Application -> Blast -> Create a local protein database file -> 选中种子序列。

构建本地数据库
3. 开始本地 BLAST:在搜索基因组里某一基因家族成员时(如此例中的 pcaF 基因)常用到本地批量序列比对,而 BioEdit 提供了方便友好的本地 BLAST 比对功能。
Ctrl+A 先选中泛基因组所有基因,接着 Accessory Application -> Blast -> Local BLAST,在弹出的窗口中选择比对算法 tblastn(此例中为将种子序列翻译为核酸序列比对泛基因组的基因),选择刚建立的数据库,选择 E 值为 1E- 5(通用的阈值,表示 100000 条比对结果中只允许 1 条是错误的),输出格式选 m8 表格形式,开始比对。
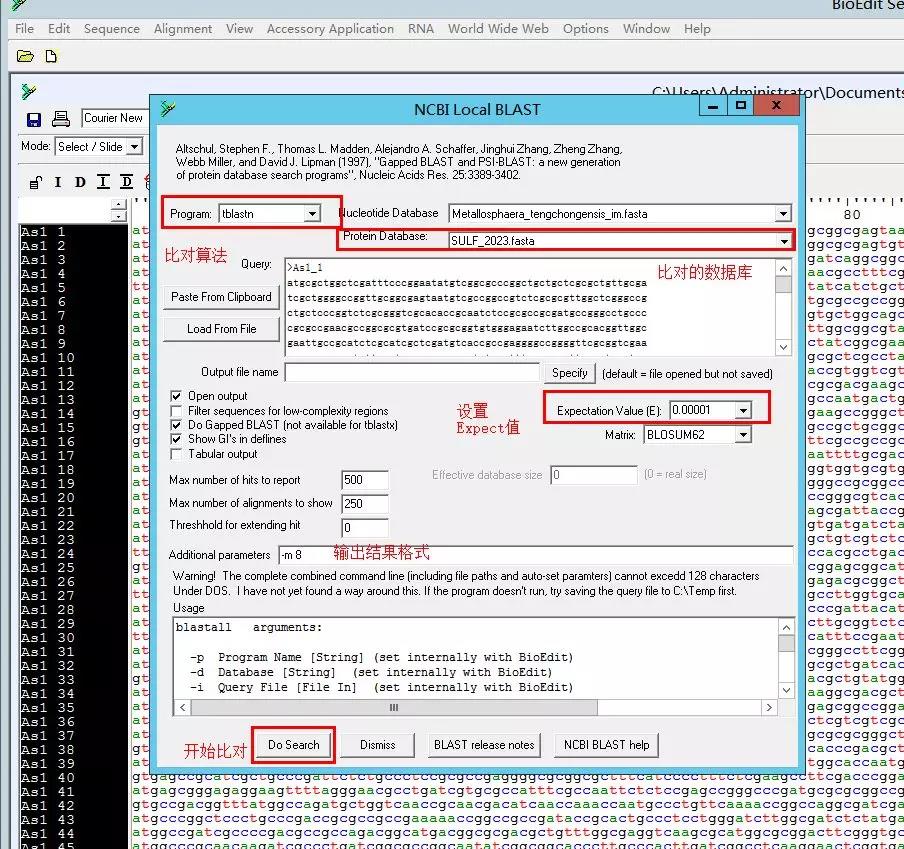
本地 BLAST
4. 提取比对上的序列:保存结果,并用 Excel 打开。找到比对出的基因序列号,复制粘贴到 BioEdit 里面查找:Edit -> Search -> Search for and select from a list of titles -> crtl+V(粘贴比对出的基因序列号)-> OK(开始搜索)
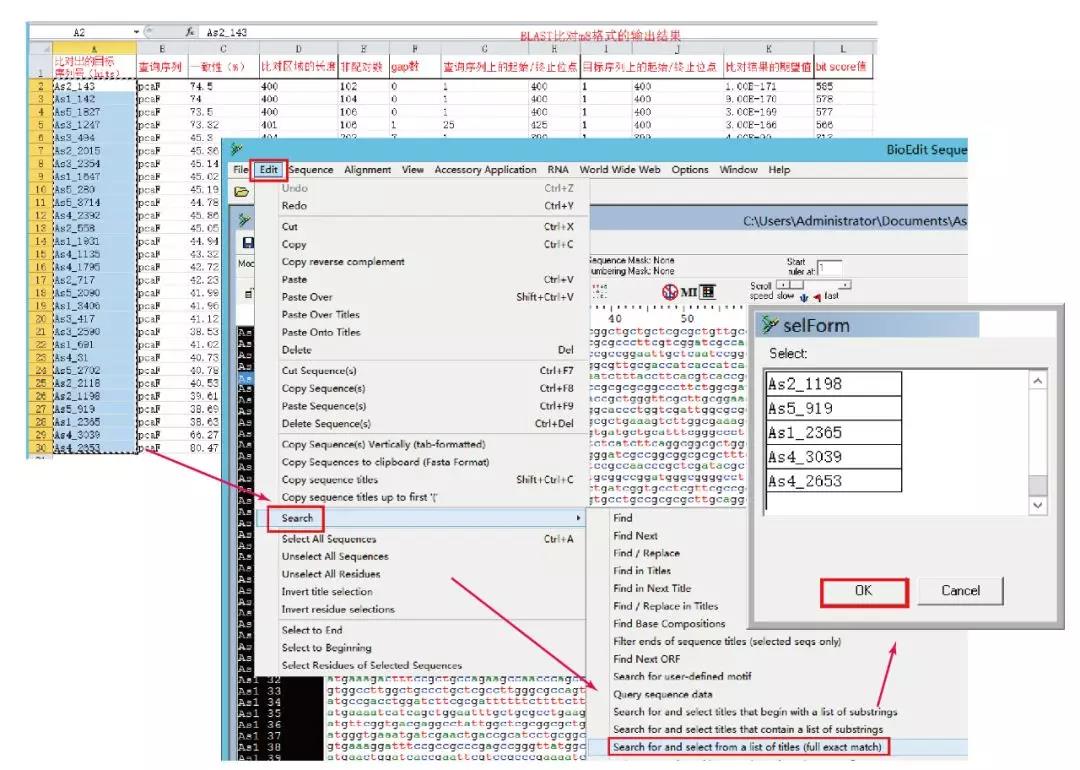
搜索比对上的序列
5. 搜索结束之后,进行 Crtl+F8(BioEdit 复制序列)-> Crtl+N(BioEdit 新建文本)-> Crtl+F9(BioEdit 粘贴序列),这样比对出的基因序列就被提取到一个新的文件里面了。
提取比对上的序列
6. 将提取出来的基因序列全部翻译成蛋白质序列并进行多序列联配(Multiple Sequence Alignment):Crtl+A 全选 -> Sequence -> Translate or Reverse-Translate –> Accessory Application -> ClustalW Multiple Alignment,还可以修改多序列联配的配色为自己喜欢的样式。
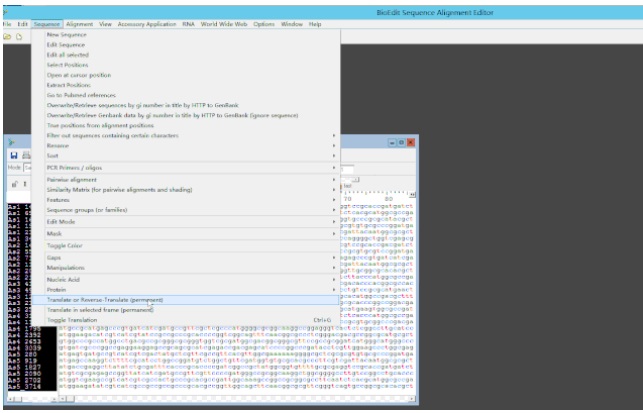
翻译以及多序列联配
7. 可以方便的预测编码蛋白的氨基酸组成、摩尔原子质量及疏水性:Sequence -> Protein
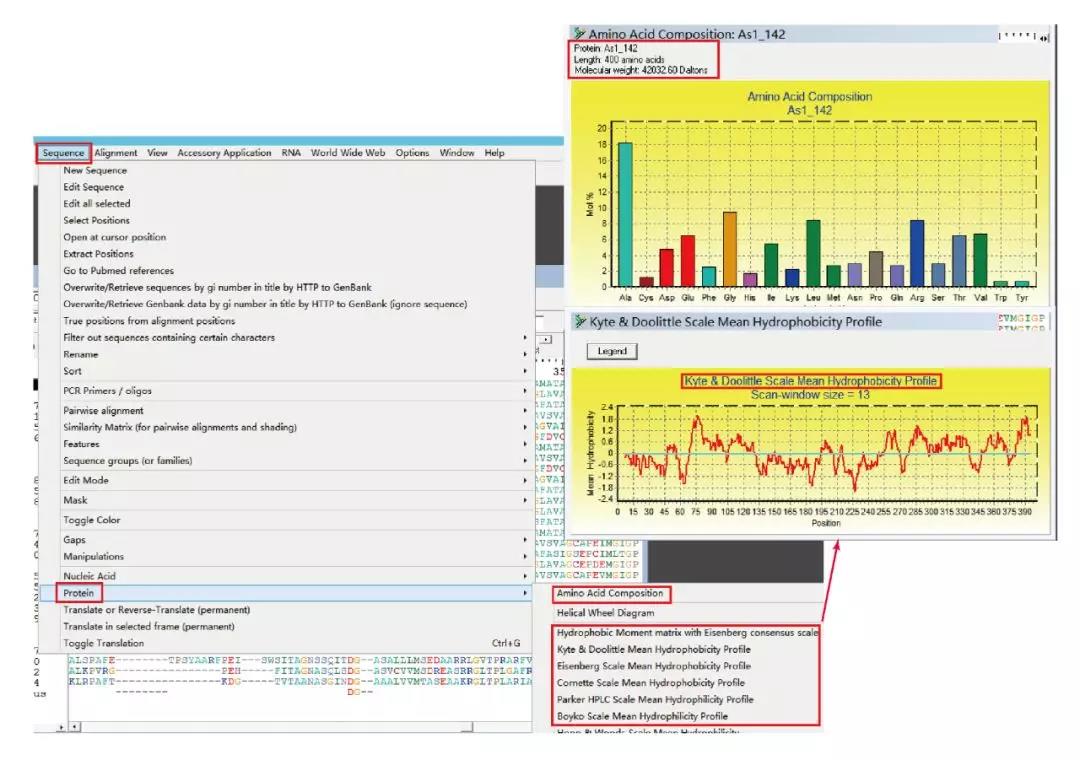
编码蛋白性质预测
8. 基因家族进化压力:提取出来的 pcaF 基因序列上传到 Ka/Ks Calculation tool 服务器(http://services.cbu.uib.no/tools/kaks),可以分析得出各分支的进化压力情况以及 Ka/Ks 值。

基因家族进化压力分析
批量计算序列 G+C 含量
1. 使用 BioEdit :选中要计算的序列 –> Sequence –> Nucleic Acid –> Nucleotide Composition,就会弹出 G+C 含量计算结果窗口,但是此方法不方便统计结果。

使用 BioEdit 计算序列 G+C 含量
2. 使用 Excel:打开 BioEdit 导出的制表符(Tab)分隔形式文本:其中 B1 单元格是要计算 G+C 含量的第一条序列,在 C1 单元格输入嵌套函数 = LEN(SUBSTITUTE(SUBSTITUTE(B1,"A",""),"T",""))/LEN(B1),它嵌套了计算字符串长度的 LEN 函数以及删除 A、T 字符的 SUBSTITUTE 函数。
分子上是替换 B1 单元格的 A 以及 T 字符,计算留下的 G+C 字符串长度,分母即计算序列总长。之后用 Excel 的智能填充功能批量计算剩下序列的 G+C 含量。
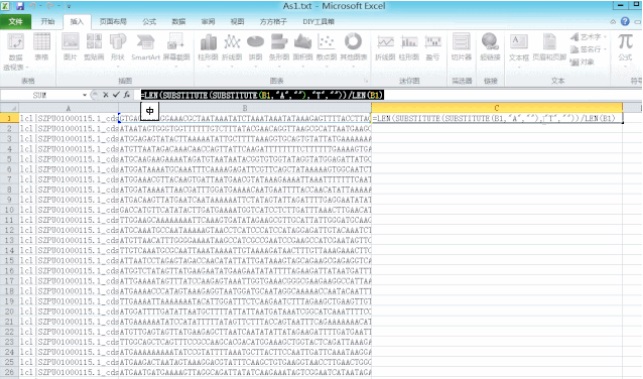
使用 Excel 嵌套函数批量计算序列 G+C 含量
以上具体实例从不同的方面介绍了软件 BioEdit + Excel 组合的强大数据处理能力,相信通过大家的探索和尝试,这些强大的软件在以后的数据处理上带给我们更多惊喜和便利!
BioEdit 下载链接:http://www.mbio.ncsu.edu/bioedit/page2.html