简介
分子系统发生分析是基于核酸或蛋白质的序列信息,通过这些信息来重建系统发育关系,推断生物进化历史。
原理
分子系统发生分析的基本原理是基于核酸或蛋白质的序列信息,通过这些信息来重建系统发育关系,推断生物进化历史。如果两条序列间相似程度越高,它们间的进化距离就越小,从共同祖先分歧的时间就越晚。反之,两条序列间差异越大,进化距离也越大,从共同祖先分歧的时间就越早。
材料与仪器
步骤
常用的分子系统发生分析软件有 MEGA、Phylip。
(一)MEGA(版本号 5.22)构建 NJ 树
A. 输入文件格式准备。构建进化树的第一步是进行多序列比对,比对后的结果导入 MEGA 中才能进行分析。
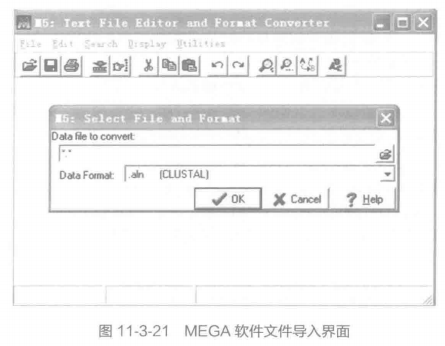
MEGA 支持的输入文件格式为 meg, 可以通过 MEGA 菜单 File 中的"Convert File Format to MEA... "将 aln、phylip、nexus 等其他格式的文件转换为可用的 MEGA 格式,如图所示。MEGA 中也整合了多序列比对软件 Clustal W, 可以直接进行比对,并保存为 meg 格式。
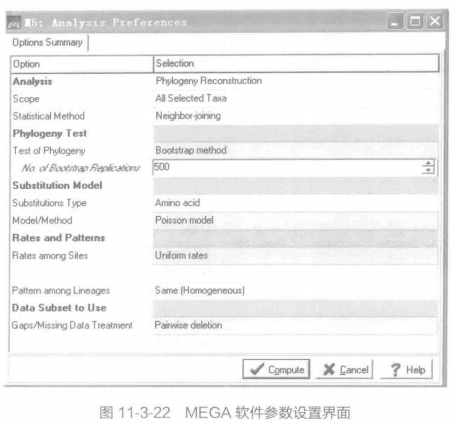
在参数窗口中还可以选择进行 Bootstrap 验证。

(二)Phylip 进化树分析
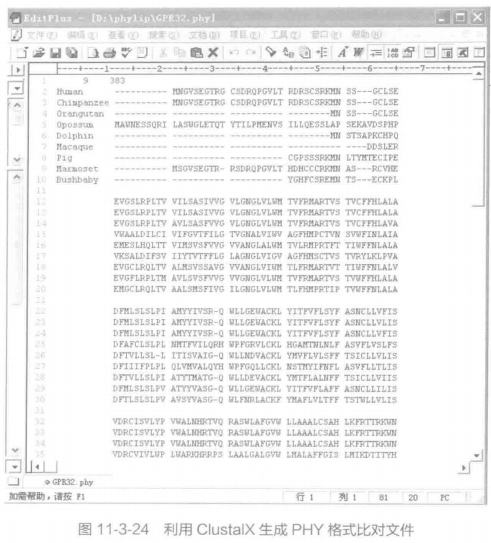
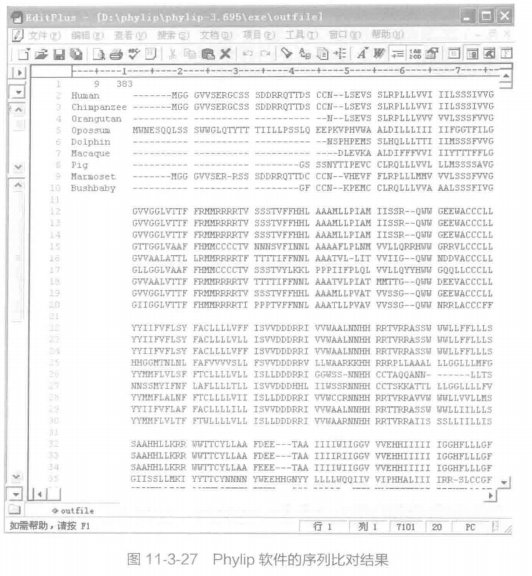
注意事项:图中的 9 和 383 分别表示 9 个序列和每个序列有 383 个氨基酸。
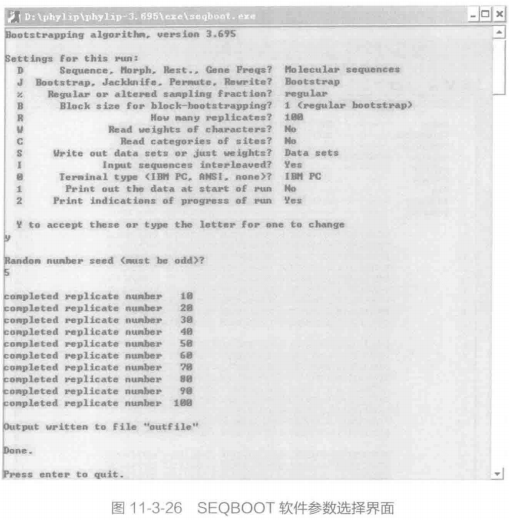
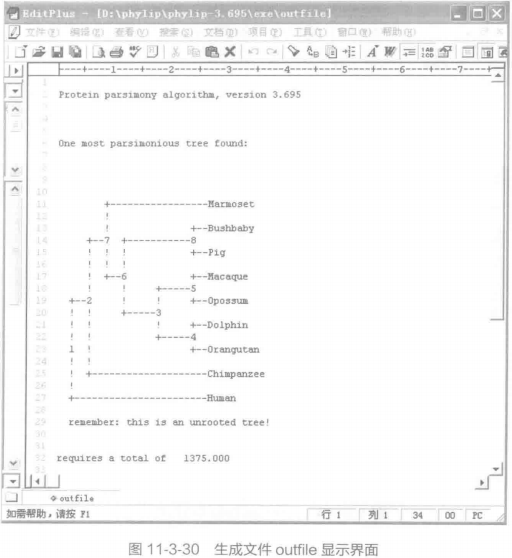
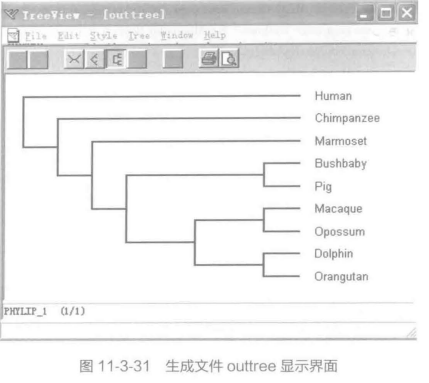
B. 打开软件 SEQBOOT,如图 11-3-25。按路径输入刚才生成的 GPR32.phy 文件后,会出现一些设置选项, 输入 Y 并回车代表你接受所有默认设置,否则你可以输入相应字母来更改设置,如图 11-3-26 所示。默认用 Bootstrap 法对进化树进行评估,R 选项让使用者输入 Replicates 的数目。当我们设置好条件后,键入 Y 按回车,并在 Random number seed (must be odd) ? 的下面输入一个"4N+ 1"的数字后,得到一个输出文件"outfle",回车后原程序结束自动关闭。所有 Phylip 程序默认的输入文件名为 infile, 输出文件名为 outfile。如果在 exe 文件夹里找不到默认的输入文件,会提示 can'tfindinputfile "infile"。outfile 打开如图 11-3-27 所示。这个文件包括了 100 个 Republicates。将刚才生成的" outfile"文件更名为"infile", 运行 protpars, 如图 11-3-28 所示。根据具体情况对相关参数进行设置,具体做法与上面类似。若选择默认设置,则输入 Y, 回车即可,如图 11-3-29。生成两个文件 outfile 和 outtree,如图 11-3-30 和 11-3-31。可以看出两个树是一样的。 但在 oufile 的树上的数字表示该枝条的 Bootstrap 支持率。到现在 9 个序列的进化树分析已经完成。


注意事项
1. 从 MEGA 官网上下载文件 MEGA5.22 Setup.exe。运行此文件,按照默认的提示进行安装,如图:
2. Phylip 是一个免费的系统发生分析软件包, 由华盛顿大学遗传学系开发。适合绝大多数操作系统。Phylip 的功能极其强大,主要包括以下几方面的功能软件: DNA 和蛋白质序列数据的分析软件; 序列数据转变成距离数据后,对距离数据分析的软件; 对基因频率和连续的元素分析的软件; 把序列的每个碱基/氨基酸独立看待时,对序列进行分析的软件; 按照 DOLLO 简约性算法对序列进行分析的软件; 绘制和修改进化树的软件。官方网址为: http : //evolution.genetics.washington.edu/ phylip.html。目前的最新版为 PHYLIP 3.695,它的下载地址为: http : /evolution.genetics.washington.edu/phylip/getme.html。下载 Windows 版的压缩包之后,解压,doc 文件夹里是关于 Phylip 子程序的使用说明,src 文件夹里所有程序的源文件,exe 文件夹里为所有的可执行程序。2. Phylip 是一个免费的系统发生分析软件包, 由华盛顿大学遗传学系开发。适合绝大多数操作系统。Phylip 的功能极其强大,主要包括以下几方面的功能软件: DNA 和蛋白质序列数据的分析软件; 序列数据转变成距离数据后,对距离数据分析的软件; 对基因频率和连续的元素分析的软件; 把序列的每个碱基/氨基酸独立看待时,对序列进行分析的软件; 按照 DOLLO 简约性算法对序列进行分析的软件; 绘制和修改进化树的软件。官方网址为: http : //evolution.genetics.washington.edu/ phylip.html。目前的最新版为 PHYLIP 3.695,它的下载地址为: http : /evolution.genetics.washington.edu/phylip/getme.html。下载 Windows 版的压缩包之后,解压,doc 文件夹里是关于 Phylip 子程序的使用说明,src 文件夹里所有程序的源文件,exe 文件夹里为所有的可执行程序。
来源:丁香实验







