材料与仪器
4 mol L 异硫氧酸胍
RNA提取试剂盒 Eppendorf 离心机 凝胶电泳装置 直接印溃装置 DNA 自动测序仪 水浴锅 分光光度计 研钵
步骤

一、.RNA 的分离
1.一般原则
差异显示技术成功最关键的因素就是RNA 的质量。为了最大限度减少RNA的降解应做到:
—戴手套
一使用无菌器具。所有塑料器皿需在180℃ 过夜
—使用无RNA酶的玻璃器皿。所有的玻璃器皿都须在180L过夜一所有的缓冲液都必须去除RNA酶。因此,应使用DEPC处理过的重蒸水(将2.5 ml的DEPC力口入到2.5L的重蒸水中后高压)
—冰上操作
2.组织勻浆
-
组织。操作前在-70X:冰冻组织并称重。在研钵里将浸于液氮的冰冻组织磨成粉末状。佩戴绝缘手套和防护眼镜以免受到液氮损伤。一定要将组织完全浸入到液氮中,因为此时损伤的细胞会释放出来大量RNA酶。用漏斗将研磨好的组织从研钵转移到一个50 ml的无菌塑料Falcon试管中。当液氮挥发,即往每50~100 mg的组织中加入ImlTRIzol,剧烈振荡直至混匀。等全部组织完全在TRIzol里溶解,RNA酶的活性就完全被GTC抑制。在这个阶段,溶液可保存在4C过夜。然而我们建议立即进行下一步处理直至达到完全没有RNA酶的步骤。
-
单层细胞。直接将ImlTRIzol力口入到培养皿(直径3.5 cm)裂解细胞。再将所得的溶液转移到2.0 ml的 Eppendorf管中。
-
悬浮细胞。IOOOg? 离心5 min。每0.5Xl〇6~l.OX1〇6的真核细胞中加入ImlTRIzol,剧烈振荡。
3.抽提
-
将含有RNA的TRIzol溶液室温孵育5 min〇
-
每毫升TRlzol中力口入0?2 ml氯仿。
-
振荡15s,室温静置30s, 再振荡15s。
-
将溶液移入2?0 ml的 Eppendorf管。
5.4℃15OOOg离心15 min。
4.RNA沉淀
1.离心后将上层无色水相移入一个新的Eppendorf管。
注意:避免混入中间相物质。因为这些物质中含蛋白及基因组DNA, 会影响到RNA的质量。
2.每毫升TRIzoI中加入0.5 ml异丙醇。
注意:如果预计总RNA少于50ng, 加入入0.5ul糖原作为载体。
3.振荡,室温解育lOmin。
4.4℃,12OOOg离心lOmin。
5.小心移去上清c
6.用75% 乙醇(每毫升TRIzol中加入Iml)洗涤RNA沉淀。
7.振荡。
8.在4℃ 下7500 g离心5 min。
9.小心移去上清。
10.将沉淀在空气中干燥15 min。
注意:沉淀不能过于干燥。请勿使用离心真空沉淀装置,否则沉淀将很难溶解。
11.用DEPC处理过的重蒸(馏)水溶解沉淀,
注意:如果所有的RNA都直接用于cDNA 合成,则用11 ul ddH20将其溶解。
12.测量OD260/280值,计算RNA产量。
13.保存RNA,加入0.1倍体积的3mol/LNaAc和2倍体积的无水乙醇。将RNA 分装成每份2.5ug 明置于-70℃。
5.RNA定量
1.取出部分RNA(如总RNA产量的十分之一),加入 DEPC处理过的重蒸水至终体积为0.5 ml(若使用0.5 ml的石英比色皿)或Iml(Iml的石英比色皿)。
注意:至少需要2ug的RNA。
2.测量260/280的OD值。
3.确定RNA产量和质量。
一 260OD值为1相当于40yg/ml的RNA
一 260/280的OD比值为2说明为RNA纯品
注意:260/280的OD比值小于2说明产物中混有蛋白和/或基因组DNA。特别是基因组DNA的存在将会造成假阳性结果。
6.DNA酶处理
为了防止可能的基因组DNA的污染,用不含RNA酶的DNA酶处理样品。
1.用IX的DNA酶缓冲液溶解RNA沉淀。
2.加入5U DNA 酶。
3.37℃ 下孵育15 min。
4.用DEPC处理过的重蒸(馏)水增容,如可至300ul。
5.加入等体积的酚/氯仿/异戊醇。
6.剧烈振荡。
7.在4℃ 下13OOOg离心4 min。
8.将上层水相转入一个干净的Eppendorf管,用分光光度计测量RNA样品的质和量。
9.沉淀RNA在-70℃ 保存。
7.RNA质量的控制
用1% 琼脂糖凝胶电泳检测RNA质量,EB染色。需使用髙压灭菌的缓冲液、玻璃器皿、电泳前用0.5mol/LNaOH清洗电泳槽。
一应清楚地看到18S和28SrRNA条带
一如果无条带或条带染色很淡,并且绝大多数EB染色都位于凝胶底部,说明RNA发生降解,不能用于差异显示
二、cDNA 合成
1.一般原则
每种RNA使用4种不同的引物[即(E)T12MA,(E)T12 MG,(E)T12MT,(E)T12MC]进行4种不同的 cDNA合成反应。而且,每一种引物都设一阴性对照。
因此,每一种RNA样品需要有8个cDNA合成反应。
2.RNA变性
1.加入2.5fxg总RNA和DEPC处理过的重蒸(馏)水至总体积为10ul。
2.根据DD类型,加入2ul(25umol/L)引物。
3.混匀。
4.70℃ 下孵育lOmin。
5.直接放置在冰上。
注意:70℃ 下孵育将破坏mRNA 分子的三级结构。若将试管从70X:移至室温,则RNA会再次退火,减少全长cDNA 分子的合成。
6.在冰上加入:
—4 ul 5X first strand缓冲液
—2 ul 0.1mol/L DTT
一I ul dNTPs(10 mmol/L)
—共19 ul
7.混匀并将试管放入25℃ 水浴。
8.孵育lmin。
9.加入1 ul反转录酶(200单位)或1 ul重蒸水作为对照。
10.混匀、孵育lOmin。
11.将试管放入42℃ 水浴。
12.再孵育50 min。
13.在70℃ 下孵育 IOmin使反转录酶热失活。
14.离心10s,将附于试管上的凝集水去除。
15.在4℃ 下保存cDNA样品
三A.DD-PCR
1.一般原则
在所有与PCR相关的移液步骤中使用带滤膜的枪头。
2.方案
用重蒸水将20fxlcDNA样品稀释至100 Ml。在PCR 反应中取用4稀释液。
注意:如果是第一次操作DI>PCR 反应,则有必要优化PCR的反应条件。例如j使用一系列不同稀释度的 cDNA可确定DD-PCR反应中模板的最佳浓度。
3.PCR反应
注意:在进行操作前应设计好移液方案。
加入如下成分:
—4 ul cDNA
一4 ul 10X缓冲溶液(其中包括:Taq酶)
一2 ul Mgcl2
—2 ul dNTPs(1mmol/L)
—4 ul DIG 标记的T12MN(2.5/umol/L)
—4DD-digo(5umol/L)
—18 ul ddH2O
—2 ul Taq酶(0.2U/ul)
总共40 ul
注意:
1.有些时候10X缓冲溶液中已含有MgCl2, 在这种情况下不加MgCl2, 并且应加入20 ul ddH2〇而不是18 ul 。
2.建议事先将10x缓冲溶液、MgC12、ddH2〇及特定引物加在一起组成混合物。
3.如果温度循环器没有加热盖的话,可滴入矿物油防止挥发。
4.先预热PCR样品至 60℃,然后再加入Taq酶。在室温中,引物的退火条件并不严格。由于酶在室温中有轻度活性,会产生过多的 cDNA 片段,并使结果的重复性不好。
4.PCR 过程
一 94℃,3 min;37℃,5 min;72℃,1min
一然后:95℃,30s;38℃,2.5 min;72℃,45s; 循环 39遍
—72°C,5 min
注意:反应条件与PCR仪有关。我们的实验条件是由Biometra优化来的,建议DD反应时尝试改变这些条件。
5.凝胶电泳
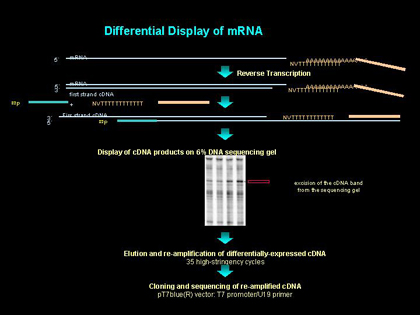
我们应用一种直接印渍装置(GATC1500), 可以将DD-PCR 产生的 cDNA 片段按照大小分离。这种仪器是专门用于分离和检测PAA 凝胶电泳的过程中直接印溃在尼龙膜上的地高新标记DNA 分子。通过抗-DIG 抗体的染色可以显示膜上的DNA片段。这种系统的优点在于无放射性。而且,它的分离范围很广,可分离长度10?800bp的片段,而经典的 PAA凝胶电泳为10~300bp或150?500bp, 因此要减少PAA凝胶的用量。因为这套操作流程专用于此套设备而设计的,且它的解说详尽清晰,这里我们只阐述一些普通的注意事项。
1.为准确比较cDNA片段,将使用相同引物扩增的PCR产物相邻点样(图 2-2)。
2.假阳性的产生是DD的一个主要问题。为避免选取到假阳性产物进行下一步分析,可增加N数,即对于每一处理组分离多个RNA样本,分别进行cDNA合成以及PCR, 电泳时使同一组的样本相邻。在凝胶上,只有处理组中每个样本都出现确定的上调或下调的 cDNA片段才予以进一步的分析。遵循这样的原则,就不会出现假阳性的结果。

三B、EDD-PCR
用ddH20将20cDNA样品稀释至25 ml,14用于PCR扩增。建议在第一次操作EDI>PCR 时,使用一系列不同稀释度的cDNA 以确定模板的最佳浓度。
注意:PCR 引物应与 cDNA合成的引物相同。例如,当用 5’-[FAM]E1T12 MG 做 cDNA合成时,也应该用 5’_[FAM]E1T12 MG 做 PCR。
1.PCR反应
试管中加入:
—I ul cDNA
一2 ul 10XPCR 缓冲溶液 II(与酶一同购买)
—1.6 ul Mgcl2(25 umol/L)
—3.5 ul dNTPs(0.5umol/L)
一2 ul 荧光 EDD 引物(2umol/L)
—2 ul BlDD 引物(2umol/L)
—0.4 ul BSA(10 mg/ml)
—7.1 ul ddH2O
—0.4 ul Amplitaq Gold(5U/ul)
总共 20ul。
注意:
1.将各组反应都用到的组分先配成混合物,包括缓冲液、MgCl2、dNTPs、BSA、H2O 以及 Amplitaq Gold。Amplitaq Gold 只有在 95”C 下孵育IOmin 才可激活,所以不用热启动。
2.对于每一对引物都设立阴性对照(加 H20)。如前所述,EDD-PCR 十分灵敏,很容易造成假阳性发生。
2.PCR 过程
一95°C,1Omin
—95℃,30s;38℃,2 min;72℃,2 min; 循环 4 遍
—95°C,30s;60t:,lmin;72℃,1.3 min; 循环30 遍
注意:这个「复合」PCR过程可以在前 5 个循环中退火温度低时(38°C), 使 BlDD 引物与许多不同的cDNA分子发生退火。与 BlDD 引物中度同源的cDNA分子会在这些条件下退火。然后,将退火温度提高至60t:就可以仅扩増最初 5 个循环中起始反应的cDNA分子,从而避免了后续的随机起始反应所造成的假阳性。
3.焚光标记的EDD-cDNA片段的凝胶电泳
我们用自动 DNA 测序仪分析了荧光 EDD-PCR产生的cDNA片段(图2-3)。采用 GATC 装置,本反应系统的优点在于不需通过放射性物质来检测 DNA分子,并且可分离长度范围很大(10~1200bp) 的片段,因而所需的PAA-凝胶也较少。使用自动 DNA 测序仪的另一个突出的优点就是可以用专门软件进行 DNA 序列自动分析,并将 EDD-数据进行数字化保存。因为自动 DNA 序列仪需要详细的操作指南,这些已经超出了本书的范围,在此就不做进一步介绍了。

4.一般原则
1.本方案中使用了非放射性标记。作为标记方法通常不会影响酶(如逆转录酶或 Tag 聚合酶)的活性,这一方案同样适用于其他包括放射性标记在内的标记物。
2.分离目的EDD-PCR 片段:
(1) 加入(32P-a)dATP, 再做 4~6 个反应循环。然后可用常规 PAA凝胶电泳分离片段。
(2) 加入DIG标记引物,再做 4~6 个循环。然后将产生的片段印溃到反应膜上。一半膜可用抗-DIG抗体染色来定位片段,另一半则切下含相应的目的片段的膜。’沸水煮膜 lOmin,洗脱 DNA,进行 20~25 个 PCR反应循环。用琼脂糖凝胶电泳鉴定分析所得片段并克隆,在这一流程中应该避免:
一用 HykmdN+, 因为这种尼龙膜结合 DNA的能力非常强,煮沸后 DNA 将不会被释放
一膜印迹后交联 DNA, 这样也会阻碍煮沸后 DNA的洗脱
来源:丁香实验