材料与仪器
SIMCA - P 多变量统计软件
[ 1H ] - NMR 抽提剂
Eppendorf 聚丙稀管 NMR 管 NMR 波谱仪
[ 1H ] - NMR 抽提剂
Eppendorf 聚丙稀管 NMR 管 NMR 波谱仪
步骤
本章描述的方法最适合于分析小麦精白面,但改进后也适用于全面粉、麦麸或其他组织,如叶和根。在开展实质等同性代谢组学实验前,如其他任何田间试验或温室试验一样,要考虑的关键步骤包括田间试验设计(见注 1) 、组织取样(见注 2 ) 和样本标记(见注 3) 。
3.1 代谢物提取和 NMR 样品准备
( 1 ) 从白面粉的每一个生物重复中,称取 3 份 30 mg ( ± 0.03 mg ) 的样品,分别装入标记好的 1.5 ml Eppendorf 管。在整个实验中,生物重复和技术重复都要随机化(见注 4) 。
( 2 ) 加入 1 ml NMR 抽提剂(见上述),盖上管盖。
( 3 ) 涡旋混匀管内内容物,直到面粉完全分散(通常大约 30 s ) ( 见注 5) 。
( 4 ) 将离心管在(50±1)℃ 准确加热 10 min。可使用聚苯乙烯浮子和预热的水浴锅来完成。离心管应放置好使内容物在水浴面下。
( 5 ) 从水浴锅中取出后,迅速将离心管转入微型离心机离心 5 min。
( 6 ) 从每管中取 800 μl 上清液,加入到干净的、标记好的 1.5 ml 聚丙烯管中。
( 7 ) (90±2)℃ 热激溶液 2 min ( 见注 6 ),如前所述,使用预热的水浴锅。
( 8 ) 将浮子从水浴锅取出后,迅速将离心管转移到管架上,置于冷库(4℃),在该温度下放置 45 min。
( 9 ) 当样品保持低温时,微型离心机全速离心 5 min。
( 10 ) 取 0.70 ml 上清液加到一个干净的标记的 5 mm 薄壁 NMR 管,加盖准备分析 ( 见注 7) 。
3.2 NMR 数据收集
( 1 ) 将 NMR 管放入 NMR 自动采样器架上,在仪器工作记录本上记录它们的位置。
( 2 ) 确认 NMR 波谱仪内可变温度单元设置为 300 K。
( 3 ) 在自动程序的样品列表输入样品细目,务必准确地输入适当的样品标记。对于输入的每个样品,选择 D2O 作为样品溶剂,扫描所需数目是 1024,参数设置为 WATERSUP ( 见注 8~10) 。
( 4 ) 开始自动程序。随后 NMR 软件自动装载每个样品进入 NMR 磁体,寻找 D2O 信号并锁定、优化信号强度 ( 通过自动匀场程序),设置接收器增益,然后收集 NMR 数据。数据收集结束前,NMR 自动程序例行自动处理数据并保存文件,随后继续下一个样品(见注 11)。
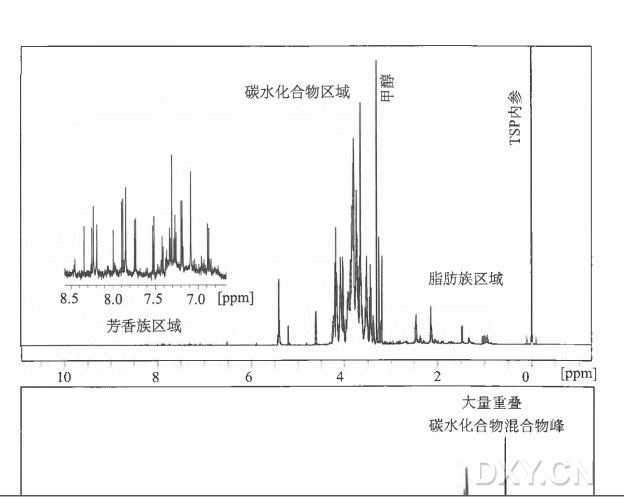
3.3 NMR 数据:肉眼检查和质量保证
( 1 ) 在整个实验结束后,打开 AMIX ( Analysis of Mixtures,Bmker Biospin,Germany) 软件,从 X Win NMR 指令选择 File > Open。在弹出窗内输入合适的存放实验数据文件的文件位置,点击 “OK”。
( 2 ) 在弹出框内选择整个实验所有样品记录,点击 “OK”。
( 3 ) 所有的 NMR 图谱此时都应在主窗口展示。检查文件确保获得满意数据( 见注 12 ) 。
( 4 ) 对步骤 3 中数据未满意的样本重新进行鉴定(见注 13)。
( 5 ) 当数据被收集和质量评估后(见注 14) ,从 NMR 管中取出样品,转移到螺帽玻璃瓶中。将这些瓶子储存在冰箱中,以备将来分析所需。
3.4 数据处理、数据库和光谱存储
在分析数据包中的数据之前,还需要对数据进一步处理。在这一步骤中,包括在 Bmker NMR 波谱数据库中( SBase) ( AMIX software 的一部分)对数据的准备。做这一步的缘由是为了确保数据集的高可比性,降低数据的复杂度,使 32k 数据点转换为含有约 1k 数据点的矩阵。由于样品 pH 的微小差异会导致某些信号中微弱化学位移差异,而 “ bucketing” 这一过程能消除位移差异所导致的数值校准问题。
( 1 ) 打开 AMIX ( Analysis of Mixtures,Bruker Biospin, Germany) 软件,选择 AMIX Tools > Prepare Data。
( 2 ) 在准备数据窗口,通过从 X Win NMR 指令选择 File > Open 打开需要加入到 SBase 的文件。在弹出窗内输入存放实验数据文件的合适位置,点击 “OK”。
( 3 ) 在弹出框内选择整个实验所有记录,点击 “OK”。
( 4 ) 所有的 NMR 图谱此时都应在主窗口展示。
( 5 ) 在 80.00 处放大 d4 -TSP 峰值。这是内标峰值。设置这一峰值为最大高度(见注 15)。
( 6 ) 重设缩放,使整个波谱宽度都能呈现。
( 7 ) 选择批处理功能,在弹出窗口选择以下选项:
( i ) 删除负峰;
( ii ) 使用用户定义范围 810.0~10.2 减噪。
( 8 ) 输入第 1 个样品的样品名(见注 16)。
( 9 ) 将这个光谱保存至光谱数据库(SBase ) 后把它关闭,输入列表中下一个光谱的样品名。
( 10 ) 将所有数据保存到 SBase 后,关闭样品准备窗口,开启 “Buckets” 窗口 ( AMIX Tools > Buckets)。
( 11 ) 选择 “Create new bucket table ”,在弹出窗口中选择 “data from SBase”。
( 12 ) 会再有一个窗口弹出。定位待 bucket 数据。
( 13 ) 在 Bucketing 选项窗口选择(见注 17)。
( i ) 待 bucketed 的数据范围(89.5~0.5 ppm)
( ii ) Bucket 宽度(0.01 ppm)
( iii ) 包容峰(所有正峰)
( iv ) 按比例缩放法(按参考区域设定比例)
( v ) 参考区域(80.05~- 0.05)
( vi ) 排除(无)
( 14 ) 用电子数据表包如 Microsoft Excel 打开 bucket 表。
( 15 ) 增加额外的行标签辅助将来数据分析(如株系、处理、时间点)。
( 16 ) 去掉对应于残留水(84.775~4.865) 和甲醇(S3.285~3.335) 的行。
( 17 ) 以 Excel 工作表形式保存文件。
3.5 多变量分析(见注 18)
( 1 ) 打开 SIMCA - P 软件(其他类似的软件包也可使用。具体的操作可能不同,但原理相同)。
( 2 ) 创建新项目(nie > New ) ,选择分析文件(由上一步创建)( 见注 19)。
( 3 ) 转置数据集(Commands >Transpose dataset ) , 使化学位移为列数据而样品为行数据。
( 4 ) 突出化学位移首行,选择该行作为主要变量 ID。
( 5 ) 突出包含样品名的列,选择该列作为主要观测 ID。
( 6 ) 突出任意描述列,将这些列赋值为定性 X 数据。
( 7 ) 继续上传数据。一旦启动,不要排除任何数值(见注 20 )。
( 8 ) 双击数据模型,选择 “ workset” (工作集)按钮。
( 9 ) 选择缩放比例选项,突出所有数据,选择缩放方法 “ ctr ”。这样通过减去平均值使数据以零为中心。
( 10 ) 通过选择变量选项和选定描述数值,排除 PCA ( 主成分分析)模型中任何定性数值。排除这些条目。
( 11 ) 自动适配并检查 PCA 模型(见注 21)。
( 12 ) 此时可以分析 PCA 得分图(见注 22) 。不同的成分图应该用不同的聚类模式分析。PC1 对 PC2 必须检测,因为这些成分常常能解释数据集当中最大的变异。
( 13 ) 根据你的描述信息(株系、处理等),利用颜色编码变量,这样便于观察数据集的趋势。
( 14 ) 对于每个得分图,应该生成两个对应的载荷图(见注 23 ) 以描述导致群集差异的代谢物。
( 15 ) 载荷图中的正负峰都能按照群集间代谢物变化赋值。这可以通过比较使用同一光谱仪在同等条件(溶剂、温度、脉冲程序)下运行正宗的纯化合物,而且采用 AMIX 在同样条件下 bucketed 收集的 NMR 频谱库实现( 见注24) 。
( 16 ) 如果没有清晰的群集,可进行判别分析。这包括在建模前设定类别到数据集 ,以及使用该信息以“强加”数据集中的差异。相应的得分和载荷图可以如前所述观察(见注 25) 。
( 17 ) 在生成载荷图的过程中,收集引起差异的数值点的信息。这只提供线索。同时也需要检查原始数据以确认代谢物的赋值和变化。载荷图的所有区域都应检查到,因为某些情况下,非常小的峰的强度变化可以比频谱中非常大的峰上的微小变化更有意义。
3.6 确定实质等同性
PCA 得分图能够快速评估样品间的相似和差异程度。载荷图用于鉴定导致差异的代谢物。目前,大多数研究 [ 2, 3 ] 认为栽培品种间的差异和环境的差异比转基因的影响更大。然而,对于导入的代谢酶或其调节物,也应考虑导入的基因对特定代谢物的直接作用。有必要建立不同的 PCA 模型,以检测环境对转基因/非转基因作物的影响。
如果去除了环境差异和品种差异后,转基因对非转基因的得分图并没有形成独立集,在分析技术所限定的范围内,认为转基因与非转基因具实质等同性。更先进的监督模型,如 PLS 或神经网络 [ 8 ] 可用于进一步探究数据集是否有差异。
3.1 代谢物提取和 NMR 样品准备
( 1 ) 从白面粉的每一个生物重复中,称取 3 份 30 mg ( ± 0.03 mg ) 的样品,分别装入标记好的 1.5 ml Eppendorf 管。在整个实验中,生物重复和技术重复都要随机化(见注 4) 。
( 2 ) 加入 1 ml NMR 抽提剂(见上述),盖上管盖。
( 3 ) 涡旋混匀管内内容物,直到面粉完全分散(通常大约 30 s ) ( 见注 5) 。
( 4 ) 将离心管在(50±1)℃ 准确加热 10 min。可使用聚苯乙烯浮子和预热的水浴锅来完成。离心管应放置好使内容物在水浴面下。
( 5 ) 从水浴锅中取出后,迅速将离心管转入微型离心机离心 5 min。
( 6 ) 从每管中取 800 μl 上清液,加入到干净的、标记好的 1.5 ml 聚丙烯管中。
( 7 ) (90±2)℃ 热激溶液 2 min ( 见注 6 ),如前所述,使用预热的水浴锅。
( 8 ) 将浮子从水浴锅取出后,迅速将离心管转移到管架上,置于冷库(4℃),在该温度下放置 45 min。
( 9 ) 当样品保持低温时,微型离心机全速离心 5 min。
( 10 ) 取 0.70 ml 上清液加到一个干净的标记的 5 mm 薄壁 NMR 管,加盖准备分析 ( 见注 7) 。
3.2 NMR 数据收集
( 1 ) 将 NMR 管放入 NMR 自动采样器架上,在仪器工作记录本上记录它们的位置。
( 2 ) 确认 NMR 波谱仪内可变温度单元设置为 300 K。
( 3 ) 在自动程序的样品列表输入样品细目,务必准确地输入适当的样品标记。对于输入的每个样品,选择 D2O 作为样品溶剂,扫描所需数目是 1024,参数设置为 WATERSUP ( 见注 8~10) 。
( 4 ) 开始自动程序。随后 NMR 软件自动装载每个样品进入 NMR 磁体,寻找 D2O 信号并锁定、优化信号强度 ( 通过自动匀场程序),设置接收器增益,然后收集 NMR 数据。数据收集结束前,NMR 自动程序例行自动处理数据并保存文件,随后继续下一个样品(见注 11)。
3.3 NMR 数据:肉眼检查和质量保证
( 1 ) 在整个实验结束后,打开 AMIX ( Analysis of Mixtures,Bmker Biospin,Germany) 软件,从 X Win NMR 指令选择 File > Open。在弹出窗内输入合适的存放实验数据文件的文件位置,点击 “OK”。
( 2 ) 在弹出框内选择整个实验所有样品记录,点击 “OK”。
( 3 ) 所有的 NMR 图谱此时都应在主窗口展示。检查文件确保获得满意数据( 见注 12 ) 。
( 4 ) 对步骤 3 中数据未满意的样本重新进行鉴定(见注 13)。
( 5 ) 当数据被收集和质量评估后(见注 14) ,从 NMR 管中取出样品,转移到螺帽玻璃瓶中。将这些瓶子储存在冰箱中,以备将来分析所需。
3.4 数据处理、数据库和光谱存储
在分析数据包中的数据之前,还需要对数据进一步处理。在这一步骤中,包括在 Bmker NMR 波谱数据库中( SBase) ( AMIX software 的一部分)对数据的准备。做这一步的缘由是为了确保数据集的高可比性,降低数据的复杂度,使 32k 数据点转换为含有约 1k 数据点的矩阵。由于样品 pH 的微小差异会导致某些信号中微弱化学位移差异,而 “ bucketing” 这一过程能消除位移差异所导致的数值校准问题。
( 1 ) 打开 AMIX ( Analysis of Mixtures,Bruker Biospin, Germany) 软件,选择 AMIX Tools > Prepare Data。
( 2 ) 在准备数据窗口,通过从 X Win NMR 指令选择 File > Open 打开需要加入到 SBase 的文件。在弹出窗内输入存放实验数据文件的合适位置,点击 “OK”。
( 3 ) 在弹出框内选择整个实验所有记录,点击 “OK”。
( 4 ) 所有的 NMR 图谱此时都应在主窗口展示。
( 5 ) 在 80.00 处放大 d4 -TSP 峰值。这是内标峰值。设置这一峰值为最大高度(见注 15)。
( 6 ) 重设缩放,使整个波谱宽度都能呈现。
( 7 ) 选择批处理功能,在弹出窗口选择以下选项:
( i ) 删除负峰;
( ii ) 使用用户定义范围 810.0~10.2 减噪。
( 8 ) 输入第 1 个样品的样品名(见注 16)。
( 9 ) 将这个光谱保存至光谱数据库(SBase ) 后把它关闭,输入列表中下一个光谱的样品名。
( 10 ) 将所有数据保存到 SBase 后,关闭样品准备窗口,开启 “Buckets” 窗口 ( AMIX Tools > Buckets)。
( 11 ) 选择 “Create new bucket table ”,在弹出窗口中选择 “data from SBase”。
( 12 ) 会再有一个窗口弹出。定位待 bucket 数据。
( 13 ) 在 Bucketing 选项窗口选择(见注 17)。
( i ) 待 bucketed 的数据范围(89.5~0.5 ppm)
( ii ) Bucket 宽度(0.01 ppm)
( iii ) 包容峰(所有正峰)
( iv ) 按比例缩放法(按参考区域设定比例)
( v ) 参考区域(80.05~- 0.05)
( vi ) 排除(无)
( 14 ) 用电子数据表包如 Microsoft Excel 打开 bucket 表。
( 15 ) 增加额外的行标签辅助将来数据分析(如株系、处理、时间点)。
( 16 ) 去掉对应于残留水(84.775~4.865) 和甲醇(S3.285~3.335) 的行。
( 17 ) 以 Excel 工作表形式保存文件。
3.5 多变量分析(见注 18)
( 1 ) 打开 SIMCA - P 软件(其他类似的软件包也可使用。具体的操作可能不同,但原理相同)。
( 2 ) 创建新项目(nie > New ) ,选择分析文件(由上一步创建)( 见注 19)。
( 3 ) 转置数据集(Commands >Transpose dataset ) , 使化学位移为列数据而样品为行数据。
( 4 ) 突出化学位移首行,选择该行作为主要变量 ID。
( 5 ) 突出包含样品名的列,选择该列作为主要观测 ID。
( 6 ) 突出任意描述列,将这些列赋值为定性 X 数据。
( 7 ) 继续上传数据。一旦启动,不要排除任何数值(见注 20 )。
( 8 ) 双击数据模型,选择 “ workset” (工作集)按钮。
( 9 ) 选择缩放比例选项,突出所有数据,选择缩放方法 “ ctr ”。这样通过减去平均值使数据以零为中心。
( 10 ) 通过选择变量选项和选定描述数值,排除 PCA ( 主成分分析)模型中任何定性数值。排除这些条目。
( 11 ) 自动适配并检查 PCA 模型(见注 21)。
( 12 ) 此时可以分析 PCA 得分图(见注 22) 。不同的成分图应该用不同的聚类模式分析。PC1 对 PC2 必须检测,因为这些成分常常能解释数据集当中最大的变异。
( 13 ) 根据你的描述信息(株系、处理等),利用颜色编码变量,这样便于观察数据集的趋势。
( 14 ) 对于每个得分图,应该生成两个对应的载荷图(见注 23 ) 以描述导致群集差异的代谢物。
( 15 ) 载荷图中的正负峰都能按照群集间代谢物变化赋值。这可以通过比较使用同一光谱仪在同等条件(溶剂、温度、脉冲程序)下运行正宗的纯化合物,而且采用 AMIX 在同样条件下 bucketed 收集的 NMR 频谱库实现( 见注24) 。
( 16 ) 如果没有清晰的群集,可进行判别分析。这包括在建模前设定类别到数据集 ,以及使用该信息以“强加”数据集中的差异。相应的得分和载荷图可以如前所述观察(见注 25) 。
( 17 ) 在生成载荷图的过程中,收集引起差异的数值点的信息。这只提供线索。同时也需要检查原始数据以确认代谢物的赋值和变化。载荷图的所有区域都应检查到,因为某些情况下,非常小的峰的强度变化可以比频谱中非常大的峰上的微小变化更有意义。
3.6 确定实质等同性
PCA 得分图能够快速评估样品间的相似和差异程度。载荷图用于鉴定导致差异的代谢物。目前,大多数研究 [ 2, 3 ] 认为栽培品种间的差异和环境的差异比转基因的影响更大。然而,对于导入的代谢酶或其调节物,也应考虑导入的基因对特定代谢物的直接作用。有必要建立不同的 PCA 模型,以检测环境对转基因/非转基因作物的影响。
如果去除了环境差异和品种差异后,转基因对非转基因的得分图并没有形成独立集,在分析技术所限定的范围内,认为转基因与非转基因具实质等同性。更先进的监督模型,如 PLS 或神经网络 [ 8 ] 可用于进一步探究数据集是否有差异。
来源:丁香实验