





4 个回答

huarenqiang5
1. workflow
进行差异表达基因分析的前提是,获取代表基因表达水平的矩阵。因此在进行分析前,必须知道基因表达矩阵是如何产生的。
在本教程中,将会简要的介绍从原始测序读数到基因表达计数矩阵过程中,所采取的不同步骤。下图是整个分析过程的流程图。
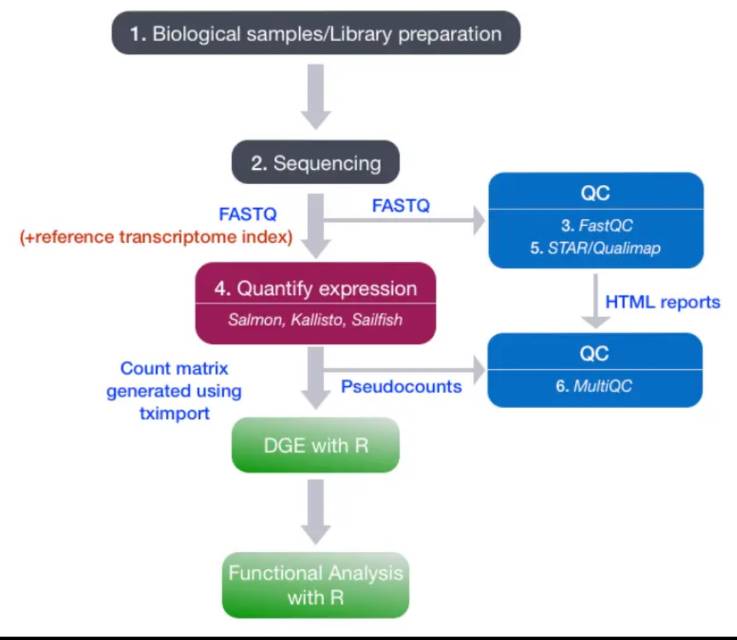
2. RNA提取与文库制备
在对RNA 进行测序前,必须从细胞环境中提取和分离出RNA 制备成cDNA 文库。下面将介绍涉及的许多步骤,其中还包括了质量检查,以确保获取高质量的RNA。
3.测序
cDNA 文库的测序将生成reads (读数)。读数对应于文库中每个cDNA 片段末端的核苷酸序列。可以选择对cDNA 片段的单端(单端读取)或片段的两端(双端读取)进行测序。
4. 质控
从测序仪获得的原始读数存储为FASTQ 文件。FASTQ 文件格式是下一代测序技术生成的序列读取的文件格式。
每个FASTQ 文件都是一个文本文件,表示样本的序列读数。每个读取由 4 行表示。
5.定量
一旦我们探索了原始读数的质量,就可以继续在转录水平上量化表达。此步骤的目标是确定每个读数来自哪个转录本以及与每个转录本相关的读数总数。
6.比对后质控
如上所述,差异基因表达分析将使用Salmon 生成的转录本/基因伪计数。然而,要对测序数据进行一些基本的质量检查,将读数与整个基因组进行比对非常重要。STAR 或HiSAT2 都能够执行此步骤并生成可用于 QC 的BAM 文件。
7.质控整合
在整个工作流程中,我们对数据执行了各种质量检查步骤。您需要对数据集中的每个样本执行此操作,确保这些指标在给定实验的样本中保持一致。应标记离群样本以供进一步调查或移除。
手动跟踪这些指标并浏览每个样本的多个HTML 报告(FastQC、Qualimap)和日志文件(Salmon、STAR)既乏味又容易出错。MultiQC ,可聚合来自多个工具的结果并生成带有图表的单个HTML 报告,以可视化和比较样品之间的各种QC 指标。如有必要,对QC指标的评估可能会导致在继续下一步之前移除样本。
一旦对所有样本执行了QC,就可以开始使用DESeq2 进行差异基因表达分析。

sswei
RNA-seq工作流程主要分为以下三步: 文库制备,使用可精确检测链方向的方法获得完整的转录组图像。 兼容FFPE RNA。 测序。 数据分析。

周末也要努力呀
参考文献:
[1] Bolger, A. M., Lohse, M., & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina Sequence Data.
Bioinformatics, btu170.
[2] Sirén J, Välimäki N, Mäkinen V (2014) Indexing graphs for path queries with applications in genome research.
IEEE/ACM Transactions on Computational Biology and Bioinformatics 11: 375–388.
[3] Kim D, Langmead B, and Salzberg SL HISAT: a fast spliced aligner with low memory requirements, Nature
methods, 2015
[4] Simon Anders, Paul Theodor Pyl, et al.(2014). HTSeq -A Python framework to work with high-throughput
sequencing data. Bioinformatics.
[5] Robinson MD, McCarthy DJ and Smyth GK (2010). edgeR: a Bioconductor package for differential
expression analysis of digital gene expression data. Bioinformatics 26, 139-140
[6] Anders, Simon, and Wolfgang Huber. "Differential expression analysis for sequence count data." Genome
biol11.10 (2010): R106

高山云初
1.去接头。首先拿到二代测序(一般是双端)cDNA序列原始文件,需要进行去接头和质量评估,这里质量评估建议用fastqc来做,不做具体介绍,去接头我用的是Trimmomatic软件,于去除Illumina平台的FASTQ序列中的Adapter,根据碱基质量值修整FASTQ序列文件。
2.构建参考基因组库。比对之前,需要构建参考基因组的索引,这里用到了Hisat2软件进行比对,所以需要用Hisat2进行索引构建。
3.将reads比对到参考基因组。这里会生成内存较大的sam文件,需要用samtools软件进行sam到bam的转换得到bam文件之后,还需对bam文件进行sort。
4.定量 使用featureCounts进行转录本的表达定量。


相关产品推荐





相关问答

