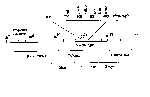Appendix G: Spectrophotometry
互联网
<center> <br /> Figure G.1 Electromagnetic Radiation Spectrum</center>
<center> <br /> Figure G.2. Schematic light transmission</center>
<center> <br /> Figure G.3. Use of the Spec 20</center>
A spectrophotometer or colorimeter makes use of the transmission of light through a solution to determine the concentration of a solute within the solution. A spectrophtometer differs from a colorimeter in the manner in which light is separated into its component wavelengths. A spectrophotometer uses a prism to separate light and a colorimeter uses filters.
Both are based on a simple design of passing light of a known wavelength through a sample and measuring the amount of light energy that is transmitted. This is accomplished by placing a photocell on the other side of the sample. All molecules absorb radiant energy at one wavelength of another. Those that absorb energy from within the visible spectrum are known as pigments. Proteins and nucleic acids absorb light in the ultraviolet range. The following figure demonstrates the radiant energy spectrum with an indication of molecules which absorb in various regions of that spectrum.
The design of the single beam spectrophotometer involves a light source, a prism, a sample holder and a photocell. Connected to each are the appropriate electrical or mechanical systems to control the illuminating intensity, the wavelength, and for conversion of energy received at the photocell into a voltage fluctuation. The voltage fluctuation is then displayed on a meter scale, is displayed digitally, or is recorded via connection to a computer for later investigation.
Spectrophotometers are useful because of the relation of intensity of color in a sample and its relation to the amount of solute within the sample. For example, if you use a solution of red food coloring in water, and measure the amount of blue light absorbed when it passes through the solution, a measureable voltage fluctuation can be induced in a photocell on the opposite side. If now the solution of red dye is diluted in half by the addition of water, the color will be approximately 1/2 as intense and the voltage generated on the photocell will be approximately half as great. Thus, there is a relationship between the voltage and the amount of dye in the sample.
Given the geometry of a spectrophotometer, what is actually measured at the photocell is the amount of light energy which arrives at the cell. The voltage meter is reading the amount of light TRANSMITTED to the photocell. Light transmission is not a linear function, but is rather an exponential function. That is why the solution was APPROXIMATELY half as intense when viewed in its diluted form.
We can however monitor the transmission level and convert it to a percentage of the amount transmitted when no dye is present. Thus, if 1/2 the light is transmitted, we can say that the solution has a 50% Transmittance. Note that it is always relative to a solution containing no dye.
Transmittance is the relative percent of light passed through the sample.
What makes all of this easy to use, however, is the conversion of that information from a percent transmittance to an inverse log function known as the Absorbance (or Optical Density).
The Beer-Lambert Law
- Definiton
- Absorbance: The negative log of the transmittance.
A = - log
T EQUATION G.1
This value is more useful in spectrophotometry than transmittance, because of plot of absorbance vs concentration yields a straight line. A plot of transmittance vs concentration is an exponential. The - log calculates the inverse of transmittance, so that absorbance increases with increasing concentration. Transmittance would decrease as we increased the amount of red dye in our example. The relationship of Absorbance to concentration was shown by two biochemists to follow the equation for a straight line, y = mx +b, where m is the slope of the line and b is the y intercept. If the measurement is made in such a way that b = 0 (that is, a solution containing no dye has no absorbance), and if we substitute Absorbance for y, concentration for x, and variant for m, we arrive at the formulation of the Beer-Lambert Law:
A =
C
where A = absorbance
C = concentration
= the extinction coefficient
Note that variant is equal to the slope of the straight line which will result from a plot of absorbance (y axis) vs concentration (x axis).
To use a spectrophotometer it is necessary to establish a known series of dilutions containing known quantities of a solute. One of these will contain no solute and is known as the blank . It is used to adjust the instrument to read 100% transmittance or 0 absorbance. In use, a 0% transmittance value (infinite absorbance) is established by placing a curtain between the light source and the photocell. Electronic control is then exerted so that the meter will read 0% Transmittance on its scale. The blank sample (containing no solute or dye) is inserted, the curtain opened and the meter readjusted to read 100% transmittance. All other measures are then made by merely inserting the samples into the light path and measuring the % transmittance. Most spectrophotometers have a built in means of direct conversion of this reading to absorbance.
After recording the absorbance for a series of standards, a plot is made of the absorbance value (y axis) vs the concentration (x axis). The slope of the line is the extinction coefficient.
Note that this may be computed directly by rearrangement of the Beer-Lambert law to
= A/C EQUATION G.2
This value can be calculated for each reading and the average taken as the value of variant. Remember that this value is a constant. Thus, once calculated, it can subsequently be used to determine an unknown concentration by one more rearrangement of the Beer-Lambert law
C = A/
EQUATION G.3
Any measured value of A can be readily converted to a corresponding concentration merely by dividing the absorbance by .
The use of the Beer-Lambert law is easy to visualize with red food coloring. It is not as easy to visualize, but none the less, just as accurate to measure wavelengths of light which are not visible. Either infra red or ultraviolet can be used. UV is more useful to biologists since many molecules (all proteins and nucleic acids) absorb ultraviolet light. The only changes that need to be made is the use of quartz cuvettes instead of glass tubes. Glass absorbs UV light and thus is inappropriate for use in a UV spectrophotometer. An instrument capable of using visible light (usually with a tungsten or halogen lamp source) and UV light is known as a UV/Vis Spectrophotometer.
OPERATION OF B&L SPEC. 20
The most commonly encountered spectrophotometer is one manufactured by Bausch and Lomb and known as the Spec 20. The 20 refers to the band size of light that it is capable of producing. If the instrument is adjust to a wavelength of 730, for example, it actually transmits light from 720 nm to 740 nm. Thus, it is not as precise or refined as instruments designed for research purposes where the wavelength may be controlled to a fraction of a nanometer. It is, however, the standard workhorse instrument found in nearly every lab.
-
Turn on the spectrophotometer and allow 10 minutes for warm up of the instrument before use.
-
Adjust the wavelength to that specificied for the procedure you are using.
-
Be sure the cover is closed on the cuvette holder and use the left knob on the front panel to adjust the dark current such that the meter is reading 0 transmittance. At this point, you are simply adjusting the internal electronics of the instrument to blank out any residual currents. This adjusts the lower limit of measurements. It establishes that no light is equivalent to 0 transmittance or infinite absorbance.
-
Insert a clean cuvette containing the blank into the holder. Be sure that the tube is clean, free of finger prints and that the painted line marker on the tube is aligned with the mark on the tube holder. Close the top of the tube holder. The blank for this exercise is the solution containing no dopachrome, but all other chemicals. The amount of solution placed in the cuvette is not important, but is usually about 5 ml. It should approximately reach the bottom of the logo printed on the side of the cuvette.
-
Adjust the meter to read 100% transmittance, using the right knob on the front of the instrument. This adjusts the instrument to read the upper limit of the measurements and establishes that your blank will give a reading of 100% transmittance (0 absorbance).
-
Remove the blank from the instrument and recheck that your 0 transmittance value has not changed. If it does, wait a few minutes for the instrument to stabilize and redo steps 1-5. Periodically throughout the exercise, check that calibration of the instrument is stable by re-inserting the blank and checking that the 0 and 100% T values are maintained.
-
To read a sample, simply insert a cuvette holding your test solution and close the cover. Read the transmittance value directly on the scale.
- Record the % transmittance of your solution, remove the test tube cuvette and continue to read and record any other solutions you may have.
It is possible to read the absorbance directly, but with an analog meter (as opposed to a digital read out), absorbance estimations are less accurate and more difficult than reading transmittance. Absorbance can be easily calculated from the transmittance value. Be sure that you note which value you measure!
ABSORPTION SPECTRUM:
Analysis of pigments often requires a slightly different use of a spectrophotometer. In the use of the instrument for determination of concentration (Beer-Lambert Law), the wavelength was pre-set and left at a single value throughout the use of the instrument. This value is often given by the procedure being employed, but can be determined by an analysis of the absorption of a solution as the wavelength is varied.The easiest means of accomplishing this is to use either a dual beam spectrophotometer or a computer controlled instrument. In either event, the baseline must be continously re-read as the wavelength is altered.
To use a single beam spectrophotometer (such as the Spec 20), the machine is zeroed first, the wavelength is set, the blank is adjusted and then the sample is inserted and read. The wavelength is then adjusted up or down by some determined interval, the zero is checked, the blank re-inserted and adjusted, and the sample re-inserted and read. This procedure continues until all wavelengths to be scanned have been read.
In this procedure, the sample remains the same, but the wavelength is adjusted. Compounds have differing absorbtion coefficients for each wavelength. Thus, each time the wavelength is altered, the instrument must be recalibrated.
A dual beam spectrophotometer divides the light into two paths. One beam is used to pass through a blank, while the remaining beam passes through the sample. Thus, the machine can monitor the difference between the two as the wavelength is altered. These instruments usually come with a motor driven mechanism for altering the wavelength, or scanning the sample.
The newer version of this procedure is the use of an instrument which scans a blank, and places the digitalized information in its computer memory. It then rescans a sample and compares the information from the sample scan to the information obtained from the blank scan. Since the information is digitalized (as opposed to an analog meter reading), manipulation of the data is possible. These instruments usually have direct ports for connection to personal computers, and often have built in temperature controls as well. This latter option would allow measurement of hanges in absorbtion due to temperature changes (known as hyperchromicity). These in turn can be used to monitor viscosity changes, which is related to the degree of molecular polymerization with the sample. For instruments with this capability, the voltage meter scale has given way to a CRT display, complete with graphics and built in functions for statistical analysis.
A temperature controlled UV spectrophotometer capable of reading several samples at pre-programmed time intervals is invaluable for enzyme kinetic analysis. An example of this type of instrument is the Beckman DU-70.
SPECIFIC PROCEDURES:
For routine use, substances to be monitored by spectrophotometry are often reacted with dyes to form a complex that is of another color, usually one easily read within the visible light range, and with precision by an instrument such as the Spec 20.EXERCISE G.1 BRADFORD PROTEIN ASSAY
MATERIALS
- Lyophilized bovine plasma gamma globulin or bovine serum albumin (BSA)
- Coomasie Brilliant Blue 1
- 0.15 M NaCl
- Spectrophotometer and tubes
- Micropipettes
PROCEDURE (STANDARD ASSAY, 20-150 µ g protein; 200-1500 µ g/ml)
-
Prepare a series of protein standards using BSA diluted with 0.15 M NaCl to final concentrations of 0 (blank = NaCl only), 250, 500, 750 and 1500 µ g BSA/ml. Also prepare serial dilutions of the unknown sample to me measured.
-
Add 100 µ l of each of the above to a separate test tube (or spectrophotometer tube if using a Spec 20).
-
Add 5.0 ml of Coomasie Blue to each tube and mix by vortex, or inversion.
-
Adjust the spectrophotometer to a wavelength of 595 nm, and blank using the tube from step 3 which contains 0 BSA.
-
Wait 5 minutes and read each of the standards and each of the samples at 595 nm wavelength.
- Plot the absorbance of the standards vs their concentration. Compute the extinction coefficient and calculate the concentrations of the unknown samples.
PROCEDURE (MICRO ASSAY, 1-10 µ g protein;
-
Prepare standard concentrations of BSA of 1, 5, 7.5 and 10 µ g/ml. Prepare a blank of NaCl only. Prepare a series of sample dilutions.
-
Add 100 µ l of each of the above to separate tubes (use microcentrifuge tubes) and add 1.0 ml of Coomasie Blue to each tube.
-
Turn on and adjust a spectrophotometer to a wavelength of 595 nm, and blank the spectrophotometer using 1.5 ml cuvettes.
-
Wait 2 minutes and read the absorbance of each standard and sample at 595 nm.
- Plot the absorbance of the standards vs their concentration. Compute the extinction coefficient and calculate the concentrations of the unknown samples.
EXERCISE G.2 LOWRY PROTEIN ASSAY
MATERIALS
- 0.15% (w/v) sodium deoxycholate
- 72% (w/v) trichloroacetic acid (TCA)
- Copper tartrate/carbonate (CTC)
- 20% (v/v) Folin-Ciocalteu reagent
- Bovine Serum Albumin (BSA)
- Spectrophotometer and tubes
- Micropipettes
PROCEDURE
-
Prepare standard dilutions of BSA of 25, 50, 75 and 100 µ g/ml. Prepare appropriate serial dilutions of the sample to be measured.
-
Place 1.0 ml of each of the above into separate tubes. Add 100 µ l of sodium deoxycholate to each tube.
-
Wait 10 minutes and add 100 µ l of TCA to each tube.
-
Centrifuge each tube for 15 minutes at 3,000 xg and discard the supernatant.
-
Add 1.0 ml of water to each tube to dissolve the pellet. Add 1.0 ml of water to a new tube to be used as a blank.
-
Add 1.0 ml of CTC to each tube (including the blank), vortex and allow to set for 10 minutes.
-
Add 500 µ l Folin-Ciocalteu to each tube, vortex and allow to set for 30 minutes.
-
Turn on and zero a spectrophotometer to a wavelength of 750 nm. Use the blank from Step 7 to adjust for 100% T.
-
Read each of the standards and samples at 750 nm.
- Plot the absorbance of the standards vs their concentration. Compute the extinction coefficient and calculate the concentrations of the unknown samples.
NOTES
The Lowry method depends on the presence of tyrosine within the protein to be measured. The standard protein must contain approximately the same number of tyrosine residues as the sample, or the procedure will be inaccurate. If there are no tyrosine residues in the sample to be measured, the Lowry method of protein determination is useless, and use should be made of the Bradford assay. In general, the Bradford assay is the method of choice for protein determinations.
EXERCISE G.3 BIURET PROTEIN ASSAY
MATERIALS
- Biuret Reagent
- Bovine serum albumin (BSA)
- Spectrophotometer and tubes
PROCEDURE
-
Prepare standard dilutions of BSA containing 1, 2.5, 5.0, 7.5 and 10 mg/ml protein. Prepare serial dilutions of the unknown samples.
-
Add 1.0 ml of each of the standards, each sample, and 1.0 ml of distilled water to separate tubes. Add 4.0 ml of Biuret reagent to each tube. Mix by vortex.
-
Incubate all of the tubes at 37 ° C for 20 minutes.
-
Turn on and adjust a spectrophotometer to read at a wavelength of 540 nm.
-
Cool the tubes from Step 3, blank the spectrophotometer and read all of the standards and samples at 540 nm.
- Plot the absorbance of the standards vs their concentration. Compute the extinction coefficient and calculate the concentrations of the unknown samples.
NOTES
The Biuret reaction was one of the first for the determination of protein concentration. It remains as a rapid determination, but is not very accurate. It is useful during protein separation procedures since there are fewer salt interference reactions than with the Bradford or Lowry techniques. The color formed is stable for about 1-2 hours and consequently all spectrophotometer readings must be made as soon as possible after the incubation step.