Using NCBI BLAST
互联网
- Abstract
- Table of Contents
- Figures
- Literature Cited
Abstract
BLAST is the most widely used software in bioinformatics research. Its main function is to compare a sequence of interest, the query sequence, to sequences in a large database. BLAST then reports the best matches, or ?hits,? found in the database. This simple program has two primary applications. First, if the function of the query sequence is unknown, it may be possible to infer its function based on the recognized functions of similar sequences. Second, if the researcher has a query sequence with a known function, it may be possible to identify sequences in the database that have similar functions. The utility of BLAST therefore depends on the researcher's choice of query sequence and database. An appreciation for the functions and limitations of BLAST are vital to using this program effectively. This unit will introduce the basic concepts behind BLAST, walk through BLAST searching protocols, and interpret common results. Curr. Protoc. Essential Lab. Tech. 1:11.1.1?11.1.36. © 2009 by John Wiley & Sons, Inc.
Keywords: BLAST; sequence alignment; sequence analysis; sequence annotation
Table of Contents
- Overview and Principles
- Strategic Planning
- Basic Protocol 1: Selecting a Sequence Using Entrez
- Basic Protocol 2: Search a Nucleotide Database Using a Nucleotide Query: Nucleotide BLAST (BLASTN)
- Basic Protocol 3: Search a Protein Database Using a Protein Query: Protein BLAST (BLASTP)
- Basic Protocol 4: Search a Protein Database Using a Translated Nucleotide Query: BLASTX
- Basic Protocol 5: Search a Translated Nucleotide Database Using a Protein Query: TBLASTN
- Basic Protocol 6: Search a Translated Nucleotide Database Using a Translated Nucleotide Query: TBLASTX
- Support Protocol 1: Preparing a Sequence in FASTA Format
- Support Protocol 2: Formatting a Sequence in GenBank/GenPept
- Understanding Results
- Troubleshooting
- A Practical Example
- Literature Cited
- Figures
- Tables
Materials
Figures
-
Figure 11.1.1 Example alignments. (A ) The two sequences differ in the second column by the change of an Ala to an Arg. (B ) The two sequences differ in the second column by the change of an Ala to a Ser. (C ) The Cys in the third column of the first sequence is aligned with a gap in the second sequence. This could be caused either by an insertion in the first sequence or by a deletion in the second. View Image -
Figure 11.1.2 The Blosum62 scoring matrix used by default by BLAST to align two protein sequences. The score for aligning two amino acids can be found by the intersection of the rows and columns of the matrix. Positive numbers mean that the two amino acids substitute one another frequently; negative numbers mean that the two amino acids rarely substitute one another. The score of an alignment can be calculated by summing the scores of each column of the alignment. Using this matrix the alignment in Figure A has a score of 18 (V‐V = 4; C‐C = 9; A‐R = −1; G‐G = 6), while that in Figure B has a score of 20 (V‐V = 4; C‐C = 9; A‐S = 1; G‐G = 6). View Image -
Figure 11.1.3 Screenshot of the NCBI Web page (http://www.ncbi.nlm.nih.gov/) showing the databases available for an Entrez search. View Image -
Figure 11.1.4 Screenshot of the BLAST program selection homepage. View Image -
Figure 11.1.5 Screenshot of the nucleotide BLAST form. View Image -
Figure 11.1.6 Example screenshot of the advanced options available under “Algorithm parameters” for a BLASTN search. The page for the other programs is slightly different, but the parameters discussed in this unit are the same for all BLAST programs. View Image -
Figure 11.1.7 Example screenshot of a BLAST results page. View Image -
Figure 11.1.8 Screenshot of the protein BLAST form. View Image -
Figure 11.1.9 Screenshot of the BLASTX form, showing the genetic codes available for the query sequence. View Image -
Figure 11.1.10 Screenshot of the TBLASTN form. View Image -
Figure 11.1.11 Screenshot of the TBLASTX form, showing the genetic codes available for the query sequence. View Image -
Figure 11.1.12 A FASTA file containing the alignment of three protein sequences. Note that some sequences have hyphens (‐) to indicate gaps. Each sequence begins with a line started by a greater than symbol (>). View Image -
Figure 11.1.13 A GenBank‐formatted flat file containing two sequences. Note that this format gives much more information about the sequence than the FASTA format. Each entry in a GenBank formatted file ends in a line containing only “//”. Multiple entries can be present in a single file; if that is the case, entries are separated by “//”. View Image -
Figure 11.1.14 Screenshot of the temporary page that is loaded after you submit your BLAST search. This is a dynamic page that is automatically updated until your results are ready. In this example, our search was assigned the Request ID GYUTSV9W016. The page also shows the results of a conserved domain search. View Image -
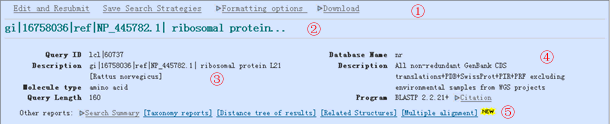
Figure 11.1.15 Screenshot of the header area of a BLAST result page. At the top are links to change how the results are displayed and to download the results. On the left, BLAST shows the ID and description of the query sequence (note that this will only be shown if you search using a GI or accession number). On the right, BLAST displays the database and program used in the search. At the bottom are links for the “Taxonomy reports” and the “Distance tree of results” pages. View Image -
Figure 11.1.16 Screenshot of the formatting options in a BLAST result page. View Image -
Figure 11.1.17 Screenshot of the Search Summary showing the different parameters used by BLAST in the search. View Image -
Figure 11.1.18 Screenshot of the “Distance tree of the results” showing a phylogenetic tree of the hits obtained in the BLAST search of the yeast formaldehyde dehydrogenase gene (SFA1) against the nr protein database. View Image -
Figure 11.1.19 Screenshot of the Graphics Summary of the BLASTP search of the yeast formaldehyde dehydrogenase gene (SFA1; accession number: NP_010113) against the protein NR database. View Image -
Figure 11.1.20 Screenshot of the Graphics Summary of the BLASTP search of the hydra tyrosine kinase HTK30 (accession number: AAC34124) against the protein nr database. View Image -
Figure 11.1.21 Screenshot of part of the Descriptions section of the BLASTP search of the yeast formaldehyde dehydrogenase gene (SFA1; accession number: NP_010113) against the protein NR database. View Image -
Figure 11.1.22 Screenshots of the alignment between the yeast formaldehyde dehydrogenase with the human alcohol dehydrogenase 5 (accession number: AAV38636.1) using different types of pairwise alignments from BLAST. (A ) Pairwise. (B ) Pairwise with dots for identity. View Image -
Figure 11.1.23 Screenshots of the query‐anchored alignment between the yeast formaldehyde dehydrogenase with its hits in Homo sapiens . (A ) Flat query‐anchored with dots for identities. The green arrow points to position 26 in the yeast query sequence; while the yeast sequence has a V at this position, all the human sequences have an I. The pink arrow points to position 37 in the yeast query sequence; the yeast sequence has an H at this position, several human sequences also have an H (represented by dots as they match the residue in the query sequence) while other human hits have a Y or a K. Note the gap between residues 27 and 28 in the yeast query sequence (black arrow). (B ) Query‐anchored with dots for identity. Note that many human sequences have the insertion of an A between positions 64 and 65 in the yeast sequence (black arrow). View Image -
Figure 11.1.24 Screenshot showing how to alter the Algorithm parameters to return 5000 hits to the query sequence. View Image -
Figure 11.1.25 Screenshot showing how to limit your BLAST search to Tetrahymena thermophila sequences in the database. View Image -
Figure 11.1.26 Graphical results of a BLAST search of the yeast formaldehyde dehydrogenase gene (accession number: NP_010113) against the protein nr database limited to the organism Tetrahymena thermophila (see Fig. ). View Image -
Figure 11.1.27 Graphical result of a BLASTP search of the Tetrahymena formaldehyde dehydrogenase protein (accession: XP_001013202) against the nr protein database. View Image -
Figure 11.1.28 Using the “Query subrange” boxes to limit the BLAST search to only part of your sequence. View Image -
Figure 11.1.29 Graphical result of a BLASTP search of the Tetrahymena formaldehyde dehydrogenase protein (accession: XP_001013202) against all the human sequences in the nr protein database. View Image
Videos
Literature Cited
| Literature Cited | |
| Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403‐410. | |
| Altschul, S.F., Madden, T.L., Schäffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI‐BLAST: A new generation of protein database search programs. Nucleic Acids Res 25:3389‐3402. | |
| Dayhoff, M.O., Schwartz, R.M., and Orcutt, B.C. 1978. A model of evolutionary change in proteins. In Atlas of Protein Sequence and Structure. Vol. 5 (M.O. Dayhoff, ed.) pp. 345‐352. National Biomedical Research Foundation. | |
| Eddy, S.R. 2004a. Where did the BLOSUM62 alignment score matrix come from? Nat. Biotechnol. 22:1035‐1036. | |
| Eddy, S.R. 2004b. What is dynamic programming? Nat. Biotechnol. 22:909‐910. | |
| Henikoff, S. and Henikoff, J. 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U.S.A. 89:10915‐10919. | |
| Korf, I., Yandell, M., and Bedell, J. 2003. Blast. O'Reilly Media, Inc. | |
| Ladunga, I. 2009. Finding similar nucleotide sequences using network BLAST searches. Curr. Protoc. Bioinform. 26:3.3.1‐3.3.26. | |
| Leonard, S.A., Littlejohn, T.G., and Baxevanis, A.D. 2006. Common file formats. Curr. Protoc. Bioinform. 16:A.1B.1‐A.1B.9. | |
| Needleman, S.B. and Wunsch, C.D. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48:443‐453. | |
| Smith, T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147:195‐197. | |
| Stover, N.A., Cavalcanti, A.R., Li, A.J., Richardson, B.C., and Landweber, L.F. 2005. Reciprocal fusions of two genes in the formaldehyde detoxification pathway in ciliates and diatoms. Mol. Biol. Evol. 22:1539‐1542. | |
| Wheeler, D. 2003. Selecting the right protein‐scoring matrix. Curr. Protoc. Bioinform. 00:3.5.1‐3.5.6. |