contamination in real-time pcr-Real-Time PCR
互联网
hi again,
still getting a signal in my template free negative control (nuclease free water instead of template)
have changed supply of nuclease free water and purchased new primers [which were diluted in the nuclease free watewr from previous experiments] and still get a signal?????
what is going wrong!?!?!?!?!?!?!
The new primers were made up in the new water right?
Diluting the old water in new water for a few 1:10 dilutions is a good way to see if the old water was the problem, as it should drop 3.3 cycles every time.
If you've changed all your reagents and you're still getting contamination, it might be something in your procedure or how you process the samples. Much more care than usual is required when doing real time as opposed to ordinary PCR.
I had similar Ct values in negative controls a few months ago. Went through the reagent change thing and so on, but still had it. Finally tracked it down to a pipettor which had a few cells of E. coli bearing my positive control plasmid on it which was used with ART tips for the real - time! :S.
real time PCR is a great technique, but it can detect anything, including nothing!
good luck
Two things...first, did you change ALL reagents including the PCR mix (possibly contaminated) and secondly, is the signal product or primer dimers? Primer dimers often result when you have no template. I am assuming your negative control is no template?...or is it no RT (reverse transcriptase)? If it is no RT and you are getting a product then you have gDNA contamination.
Hope this input helps.
I get a product very late in my PCR in my water despite changing waters, too (30-32 cycles). I asked BioRad and they said not to worry about it, that this is common in real-time....how late are your negative controls coming up?
I may get amplification after 35 cycles however when run on a gel it's usually non-specific as a very faint smear can be seen if any. If there was a distinct band of expected size I would be worried!!!
Nick
in my experience the amplification that you get in the water control, or at the very end of the run, is usually due to primer dimers. even if your primers dont form visible primer dimers (on a gel) i think the technique is sensitive enough to pick them up. you can usually see if that's what they are by looking at the melting curve. since the primer dimers are smaller than the product, they should have a lower melting point.
i wouldnt worry too much about them (if that's what they are), because the C(t) should be pretty different to your product's C(t).
the melt curve's are pretty good, and the product i'm getting in my negative controls (although a lower peak height) is at the same temperature as my samples..... a guy from biorad is coming tomorrow to look so gingers crossed...
Before any of you get too concerned about what your contamination band is:
1) you must sequence a band from your reaction (gel extract or pcr cleanup), to know what you are looking at for sure..(if the band is not that different in size from your intended amplicon.
2) reactions without a template will often, more often than not, give a band eventually. Without template, the even the most inefficient primer interactions will eventually make a product (on their own). While they may not be efficient at first, eventually the first primer dimer products will become very efficient template for primer dimer product formation. So a bona-fide negative sample can give you these bands.
3) As a rule of thumb, you can assume that primer dimer bands, or even mild true contamination, that is MORE than 5 Ct. values away from your samples/standards are ok to use. 10 Ct. would be better.
4) Make sure you start with a clean area, and try to set up your PCR away from where PCR reactions are opened (ie where gels are loaded!)
5) you can UV irradiate your tubes (open) and even your mastermix before you add dye and Taq to destroy TEMPLATE (thymidine dimers formed). this usually does not seem to harm primers or dNTPs in the mix. irradiate (5 min) your pipettors too, facing up in a beaker. Use barrier filter tips.
6) DO NOT take any template out of storage untill you have made yoru mastermixes then put all the master mix components back into storage.
7) do not be afraid to run real time reactions for far less cycles (ie. not 40), such as 25-28 cycles, if your samples are showing product build up by that time. the melt curves done at that time, and checking the reactions on a gel will show if your product is popping up long before primer dimers or mild contamination do.
good luck.
Ken Mitton, PhD
Assistant Professor of Biomedical Sciences
Oakland University Eye Research Institute
Hi, Flashboy. I EXACTLY have the same result as you. Please tell me what the BioRad engineers said.Thx a lot!!!
still getting a signal in my template free negative control (nuclease free water instead of template)
have changed supply of nuclease free water and purchased new primers [which were diluted in the nuclease free watewr from previous experiments] and still get a signal?????
what is going wrong!?!?!?!?!?!?
If the melting curve peak of the contamination is the same than in your samples it is, defenetly, a contamination, rather than primer dimers.
First, try to make new primer stock and change the reactives you use. If the contamination persists it might be on your pipets. You can wash them with NaOH 1M O/N. It is probably not very safe for your pipets to have a long life, but you will get rid off the contamination.
If it is an enviromental contamination, you better try to make the mix in another lab.
I had a lot of problems with contaminations ... hope my experience is useful for you to have a quick solution
Ah... the old contamination chestnut...
If the product in your no-template controls is amplified by your gene-specific primers, then the problem is likely to be contamination with your PCR product - follow instructions in the posts above to resolve this,
If you're using 18S rRNA as a housekeeping gene, and your unexpected product is being amplified from 18S primers, don't forget that these primers are generally 'universal' - so they will amplify the 18S gene from almost *anundefined eukaryote. Fungi are the usual suspect here. If you're using 18S primers you should autoclave and UV-irradiate your PCR tubes / plates before use. If you don't have a UV-crosslinker, you can invert a transilluminator and irradiate the (open) tubes / caps in racks underneath for 10-15 minutes or so.
good luck,
Del.
I did not know what type of dye you have used, SYBR Green or Taqman probe? If you are using SYBR Green, you may need to worry about primer dimers. If you are using the taqman probe, it dose not make sense primer dimers will show the amplification, because no amplicon will be generated by primer dimers. Amplification shows up only when the probes are broken down during amplicons synthesis.
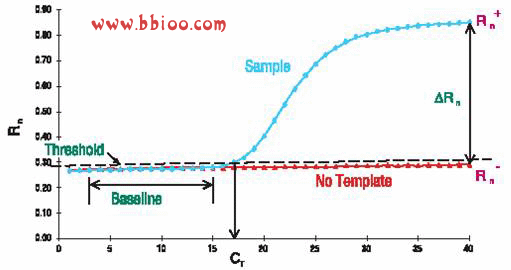
I am using ABI Prism 7000 machine I noticed that sometimes the noise may give me wrong report when threshold line was set too low. Please check the threshold line settings and amplification curve. Change the threshold line settings may also change Ct value a lot.
This paper has some interesting studies on using DNAse, UV, restriction enzymes, and 8-MOP to overcome contamination:
Has anyone here ever tried 8-MOP or DNAse treatment? The paper above describes a significant loss of sensitivity when using it 8-MOP, but very little using DNAse in combination with AvaI.
QUOTE(flashboy @ Apr 1 2005, 03:11 AM) [snapback]13331[/snapback]
still getting a signal in my template free negative control (nuclease free water instead of template)
have changed supply of nuclease free water and purchased new primers [which were diluted in the nuclease free watewr from previous experiments] and still get a signal?????
what is going wrong!?!?!?!?!?!?!
Hi flash boy,
maybe you should consider to use UNG for carry-over prevention and dUTP in your dNTP mix. Furthermore, did you check if your primers are forming dimers? Did you do a testrun of this PCR-product on an analytical agarose gel?
I hope this might give you some hints.
Cheers,
ophris
This problem is probably due to contamination of amplified products in the air from previous runs using the same primers. If your room has no windows that can be opened and is central air conditioned, I bet this is it. All you need to do is to open the windows and let the air circulate. We have been using a huge fan to blow the lab once in a while.
Also, never open a real-time PCR tube in the lab where you prepare your real-time PCR reactions. If you run an agerose gel to check your real-time PCR amplification, you contaminate the lab. It is very hard to get rid of the air contamination - it usually takes months before your negative control shows negative again.
Jing