|
Summary
Long DNA oligos containing the target sequence are cloned into LentiLox 3.7 (see protocol ). Once clones have been isolated, virus is produced by transfecting 293 cells and collecting supernatant. This supernatant is then used to infect cells of interest directly, or concentrated for use in embryo infections.
LentiLox Vector
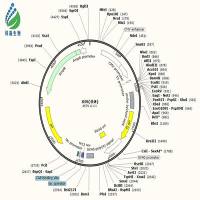
LentiLox 3.7 (see sequence and map ) is a lentiviral vector designed for inducing RNA interference in a wide range of cell types, tissues and organisms. We have use this vector to infect and efficiently silence proteins in hematopoietic stem cells and their progeny, and have used infected embryonic stem cells and single cell embryos to create transgenic animals (Rubinson and Dillon et al, Nature Genetics, 2003). These lentiviruses are pseudo-coated with VSV-G and are capable of infecting human cells, and thus present important biosafety issues. This is explained here .
Packaging Vectors
We use the 3rd generation packaging systems for lentiviral production originally published by Dull T et al (J Virol. 1998 Nov;72(11):8463-71.). These vectors include: pMDLg/pRRE (gag/pol elements), pRSV-REV, and pMD.G (env elements).
Sources of these packaging vectors include:
Invitrogen- Viral Power Lentiviral Support Kit K4970-00
Trono Laboratory, University of Geneva Medical School, Geneva, Switzerland
Naldini Laboratory, IRCC, University of Torino, Italy
Verma Laboratory, Salk Institute, San Diego, CA
Virus Production
-
Plate 12 x 106 293.T in 20 ml on a 15 cm2 plate 24 hours before transfection . In general, two 15cm plates per virus. It is essential that the cells be well-maintained and of relatively low passage number.
-
Mix the following DNAs (made w/ Endo-free Qiagen Kits) in a FACS tube. The DNAs should be in Endo-free TE at a concentration of 0.5m g/m l.
For 3 plasmid system:
20 m g vector,
10 m g VSVG
15 m g D 8.9
For 4 plasmid, system (recommended),
20 m g vector,
10 m g VSVG
10 m g RSV-REV
10 m g pMDL g/p RRE
-
Add 400 m l 1.25 M CaCl2 and 1.5 ml H2 0 and mix by tapping gently.
The following steps are done 1 plate at a time.
-
Add 2 ml of 2X HBS dropwise to DNA mixture while bubbling with a Pasteur pipette. When finished, continue to bubble for 12-15 seconds.
-
Take plate of 293T out of the incubator (plate remains in incubator for long as possible), and add transfection mixture dropwise all over the plate. Gently swirl plate from front to back, and return immediately to incubator.
-
3.5 to 4 hours later, remove media, wash 2x with 10ml warm PBS, and add 20 ml warm D10 onto plate and place in incubator.
-
36-48 hours after transfection, harvest viral supernatant and spin at 2000 rpm, 7 min at 4� in a 50ml tube.
-
Filter viral SN through .45 um filter. Add 35ml of filtered supernatant to an ultracentrifuge tube. Balance tubes with additional media. Cover tubes with small piece of parafilm . (It is useful to titer some of the leftover supernatant to determine if there is loss of virus during concentration.)
-
Spin tubes using a SW-28 rotor at 25,000 rpm, 90 min, 4 �. Decant liquid and leave tube upside down on kimwipe for 10 min. Aspirate remaining media being careful not to touch bottom of tube.
-
Add 15m l cold PBS (for embryo infections, or any volume you wish) and leave tube at 4� O/N with no shaking.
-
To resuspend , hold tube at angle and pipet fluid over pellet 20 times, being careful not to touch pellet with tip. It is expected that the pellet not be resuspended after this is complete. This pellet does not contain virus and can be discarded.
-
Aliquot or use virus. Virus should be aliquoted , flash-frozen in liquid nitrogen and stored at -80. There should be no change in titer with freezing concentrated virus. Avoid multiple freeze-thaws.
Titering Virus
-
Plate 4x105 293.T cells/well in a 6-well plate 12-24 hours prior to titering . It is helpful to have an additional well as a negative control that you mock infect with D10+polybrene but without virus.
-
Make a stock solution of D10 with 8 m g/ml polybrene .
-
Generate a 10-fold dilution series of virus in the D10+polybrene. Using 1.5mls/well you should have 1 m l, .1, .01, .001, .0001, and .00001 u L of virus/well.
-
I ncubate at 37 degrees O/N. Replace media with fresh D 10.
-
At least 48 hours after infection trypsinize cells for FACS analysis. (Trypsinize , inactivate with media, spin, and resuspend in cold PBS).
-
FACS analyze for EGFP expression and record the percentage of cells that are EGFP positive.
-
Use a well that has between .1% and 10% of cells expressing EGFP to determine titer.
Sample calculation assuming 1% infection from the well with .01 m l of virus.
.01 (percentage of cells that are EGFP positive) x 4 x 105 = 4 x 103 positive cells.
4 x 103 x 100 (dilution factor) = 4 x 105 viral particles/m l
In general you should have at least 5 x 105 viral particles/m l for embryo infections .
|