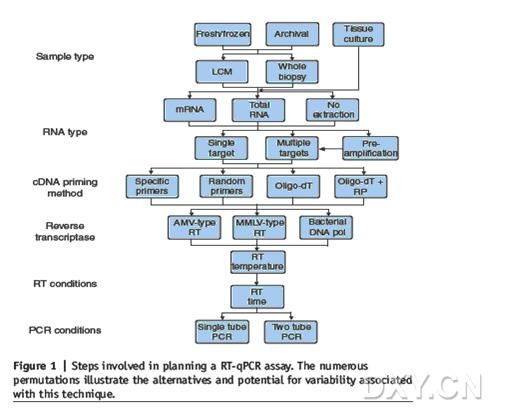
QRT-PCR
互联网
Comparison of normalisation methods
There is an ongoing debate what is the best way to normalise qPCR data. Reference genes are the most common method, although single unverified reference genes invalidate the qPCR data generated. Total RNA, ribosomal RNA, and genomic DNA have been suggested as alternative methods.
Reference genes
Most common method. Frequently, a panel is used for normalization, e.g. [1] not just a single reference gene and including data on suitability as reference genes. Often housekeeping gene is used here instead of reference gene but the term is poorly defined and can be misleading. It is to be noted that panels are often composed of genes that are supposed to be stable based on their function. However, more than 100 peer-reviewed articles report problems related to genes chosen from a panel, because they were not suitable for a particular context. A recent approach is to select a reference gene based on its stability across microarrays done within one''s condition of interest. There is a public tool called RefGenes that searches a microarray database of more than 50,000 arrays to identify genes that are stable across subsets of conditions. It is available at the Genevestigator website [2].
RNA
Total rRNA [3] [4], or total RNA. Drawback: rapidly dividing cells will have more rRNA and different rRNA/mRNA ratio which will complicate comparison; difference in cDNA synthesis not taken into account.
Genomic DNA
Genomic DNA or cell number. Drawbacks: RNA degrades faster than RNA which can distort the data; sample cannot be DNase treated; efficiency of cDNA synthesis not taken into account.
Reference mRNAs
- Main article: Choosing reference genes for qPCR normalisation
Picking reference genes will make or break your quantification via qPCR (real time PCR). If you pick only one reference gene and your pick is not constant across different conditions or samples, your results will be skewed. Choose several reference genes and check whether they satisfy the criteria for a good reference gene. Some commonly used reference genes, like 18S and GAPDH, are known to be problematic but continue to be used.
- Ajeffs 06:55, 21 April 2007 (EDT): Screen a handful of ref genes, select the most stable using genorm, bestkeeper etc, use at least 2 reference genes for subsequent reactions and normalisation. Inlcude your genorm M values when publishing qPCR data.
Primer selection
- Main article: Choosing primers for qPCR
Choosing suitable primers is an early crucial step in your qPCR experiment. Reusing a tested primer pair from a repository or publication can save you some time. Otherwise primer selection from scratch is similar to that for a standard qualitative PCR experiment but the product size is typically much smaller (below 200nt) and the amplification characteristics of the primer have to be rigorously tested.
Quantification methods
There are 3 common quantification methods. The standard curve method is the only one that gives you are absolute concentration. Both the Pfaffl method and the ΔΔCt method produce relative data with the Pfaffl method being superior.
Standard curve method
- requires template at known concentration (e.g. cDNA or TA cloned PCR product)
- requires dilution series of known template for standard curve (more wells)
- yields absolute concentrations by comparing unknown samples to known
Pfaffl method
- requires that primer efficiency be known but needs to be determined only once with a standard curve or a different method
- produces relative amount (e.g. treated is 2x untreated)
(named after the inventor; see Pfaffl 2001 PMID 11328886)
ΔΔCt (delta delta Ct)
- easiest, oldest, least reliable
- assumes that primers for unknown and reference gene have very similar efficiency
- or that v little correction is necessary (i.e. reference gene almost same level)
- yields relative amounts
(Ct = cycle threshold; point when fluorescence reading surpasses a set baseline)
Primer efficiency estimation
A Ct difference of 1 between two samples has a different meaning depending on the efficiency of the primers used. If primers are 100% efficient, then ΔCt = 1 means one sample has twice the amount of template compared to the other. The simple ΔΔCt method, described above, often wrongly assumes perfect efficiency. It is better to experimentally verify the primer efficiency and use the Pfaffl method instead. The standard method takes the primer efficiency into account via the standard curve run with each sample. However, primer efficiencies in the standard curve dilutions and the actual samples are not necessarily the same.
Linear regression on dilution curve Ct data
Primer efficiencies can be calculated by making a dilution series, calculating a linear regression based on the data points, and inferring the efficiency from the slope of the line. For a base 10 logarithm the formulae is:
- efficiency = 10^(-1/slope)
Slopes between -3.3 and -4 will thus give you estimated primer efficiencies between 100% and 78% respectively. It can happen that the calculated efficiencies are above 100% [5] [6] [7]. This may be due to incorrect template concentrations, too concentrated template, inhibition of the PCR reaction, unspecific PCR amplification, mistakes in the calculation [8], etc.
See a figure explaining the fitting process from the Hunts'' qPCR tutorial [9].
Efficiency estimation based on the kinetics of single PCR runs
Efficiency (and Ct values) can also be calculated from the fluorescence data of a single PCR run or preferably replicates of the same PCR. The Miner algorithm (PMID 16241897) is an example for this type of method and can be used online at [10].
qPCR data quality
Sources of variability: Operator
Due to the small amount of liquid handled and the sensitivity of the technique, operator variability is high. Bustin reports that the same qPCR experiment repeated by 3 people using the same reagents lead to very different copy number estimations [Bustin 2002 PMID 12200227, figure 3]:
- person A: 8・7 × 105
- person B: 2・8 × 105 different by a factor of 3!
- person C: 2・7 × 103 different by a factor of 300!!
Sources of variability: Reagent lots/age
Different lots of reagents can lead to different results. Experiment repeated by same operator 5 times, same RNA sample, different kits; values are copies/μg total RNA:
- kit 1: 13±32 × 107
- kit 2: 5.4±1.6 × 107 - different by a factor of 2.4
Similar experiment with old (9 months 4°C) and new probe (3 months 4°C), values are copies/μg total RNA:
- old: (5.6 ± 1.3) x 103
- new: (3.8 ± 0.6) x 108 - different by a factor of 100''000!!
both experiments above from [Bustin 2002 PMID 12200227, figure 4]
More information
- Partial transcript of a webcast discussing qPCR data quality
Notes
- The most commonly used specialist reverse transcriptase enzyme for cDNA production is AMV reverse transcriptase. It has RNase H activity (so that RNA molecules are only transcribed once) and has a high temperature stability (to reduce RNA secondary structure and nonspecific primer annealing) [1].
- Since RNA can degrade with repeated freeze-thaw steps, experimental variability is often seen during successive reverse transcription reactions of the same RNA sample [1].
- Reverse transcriptase enzymes are notorious for their thermal instability. Repeated removals from the freezer can degrade the efficiency of the enzyme [1].
-
Producing total cDNA from total RNA can be advantageous because
- cDNA is more stable than RNA so making total cDNA allows you to make multiple sequence-specific RNA measurements [1].
- This approach could reduce experimental variability stemming from RNA degradation [1].
-
To make total cDNA
- Use a polyT primer (most but not all eukaryotic mRNA) or random decamers (prokaryotic mRNA) [1].
- Random decamers give longer cDNAs on average than random hexamer primers [1].
- Use longer reverse transcription reaction times [1].
- Ensure that the concentration of deoxynucleotides doesn''t run out [1].