Using Evolutionary Information to Find Specificity-Determining and Co-evolving Residues
互联网
308
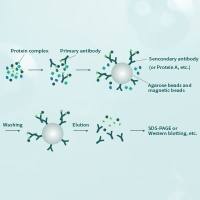
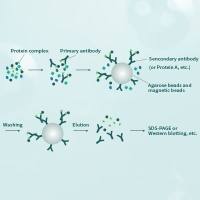
Intricate networks of protein interactions rely on the ability of a protein to recognize its targets: other proteins, ligands, and sites on DNA and RNA. To recognize other molecules, it was suggested that a protein uses a small set of specificity-determining residues (SDRs). How can one find these residues in proteins and distinguish them from other functionally important amino acids? A number of bioinformatics methods to predict SDRs have been developed in recent years. These methods use genomic information and multiple sequence alignments to identify positions exhibiting a specific pattern of conservation and variability. The challenge is to delineate the evolutionary pattern of SDRs from that of the active site residues and the residues responsible for formation of the protein’s structure. The phylogenetic history of a protein family makes such analysis particularly hard. Here we present two methods for finding the SDRs and the co-evolving residues (CERs) in proteins. We use a Monte Carlo approach for statistical inference, allowing us to reveal specific evolutionary patterns of SDRs and CERs. We apply these methods to study specific recognition in the bacterial two-component system and in the class Ia aminoacyl-tRNA synthetases. Our results agree well with structural information and the experimental analyses of these systems. Our results point at the complex and distinct patterns characteristic of the evolution of specificity in these systems.