Single Primer (Semi-Random) PCR
互联网
<center> <b><font>Single Primer ("Semi-Random") PCR</font> </b></center> <center> <p> <font>July 26, 2000 ECK</font></p> </center>
Description
Single primer PCR allows amplification from known to unknown regions in chromosomes, phage, plasmids, large PCR products and other sources of DNA.
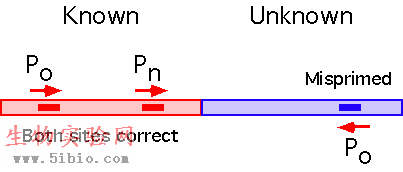
At sufficiently low stringency, any primer will misprime while continuing to bind specifically to its intended site. Conditions can usually be found allowing mispriming sufficiently close (<3.5 kb) to the correct site to permit amplification anchored at the same.
Reamplification with a nested primer and the original outside primer generates a product with unique ends. The resulting size shift can be used to diagnose the correct product, which can then be sequenced from either end.
Two methods are presented. The "Short" method can be done quickly and works about 80% of the time in our lab for any given primer Po (see figure below) It has been adapted from Hermann et al (1). The "Long" method, developed in our laboratory (2), requires more time but will often work when the short method fails. Another advantage of the long method is that the product has unique ends, allowing convergent sequencing.
When amplifying out of inverted repeats at the ends of certain elements such as Tn10 or Tn5, gel purify bands before sequencing. If you try to sequence the crude PCR reaction, you will get superimposed sequences coming out of both ends.
In our lab, PCR is always done in an Air ThermoCycler (Idaho Technologies, PO Box 50819, Idaho Falls, ID 83402), which ramps quickly, allowing short dwell times during denaturation and annealing. This enables rapid and stringent polymerization, while minimizing enzyme aging and template hydrolysis. We have not tested the protocal on other machines.
In an appendix at the end, I present some primers which have worked well in our lab when sequencing out of common insertion sequences.
Protocol
Single primer PCR works best when when there are nested primers Po and Pn with sites in the known region:
1. Short Method
When this method fails, try varying the annealing temperature in the second cycling regime. If that doesn't work, try changing primers. If problems persist, switch to the long method.
Do the following PCR reaction:
15 µL H2O
3 µL 10xM-PCRB
3 µL 4 dNTPs @ 2 mM each
3 µL Po @ 5 µM
3 µL template diluted to about 1 ng/µL
3 µL "T/TS" @ 0.4 u Taq/µL
--------
30 µL
Cycle as follows:
30"x94°C
20 cycles 0"x94°C 0"x55°C 1'x72° S=9
30 cycles 0"x94°C 0"x40°C 1'x72° S=6
30 cycles 0"x94°C 0"x55°C 1'x72° S=9
Clean up:
Add 1 µL ExoI nuclease @ 1 u/µL
1 hr x 37°
Purify using Qiaquick (or related) technology, elute in 30 µL TE
Check 3 µL on 1% agarose gel
Sequence with primer Pn
2. Long Method
Stringency optimization is done with Po, and reamplification using a biased ratio of Po and Pn.
Make 3 reactions with varying [Mg++]:
15 µL H2O
3 µL 10x H, M or L-PCRB
3 µL 4 dNTPs @ 2 mM each
3 µL Po @ 5 µM
3 µL template diluted to about 1 ng/µL
3 µL "T/TS" @ 0.4 u Taq/µL
--------
30 µL
Distribute 3 10 µL aliquots of each mix into capillaries and discard remainder. Load sets of 3 capillaries, one from each mix, consecutively into the same machine with the annealing temperature ("Ta") set at 40, 45 and 50°, respectively.
Cycle as follows:
30"x94°C 20 cycles 0"x94°C 0"x55°C 1'x72° S=9
30 cycles 0"x94°C 0"xTa 1'x72° S=6
30 cycles 0"x94°C 0"x55°C 1'x72° S=9
Load a 0.8% agarose gel according to the following pattern:
Reactions arrayed in this fashion have roughly increasing stringency from left to right.
Find the highest stringency at which distinct bands are visible. Usually, all such bands are anchored at the specific site corresponding to Po. This is especially true if there are only one or two, the situation we wish.
Isolate DNA. We generally core the band with a yellow tip, and soak it overnight at 4° in a small amount of TE or H2. The supernatant provides the template for reamplification.
It is also useful to reamplify the unfractionated products of the first amplification, after first removing primers by Qiaquick, Wizard, or similar methodology. Running the first and second amplification products side by side will reveal correctly anchored bands, as they will be shifted with respect to parental bands.
Reamplification
40 µL H2O
10 µL 10x M-PCRB
10 µL 4 dNTPs @ 2 mM each
10 µL Pn primer @ 5 µM
10 µL Po primer @ 0.2 µM
10 µL DNA from part 1
10 µL "T/TS" @ 0.4 u Taq/µL
---------
100 µL
Load glass capillaries and amplify:
30"x94°C
30-40 cycles 0"x94°C 0"x55° 30" x 72°
5' x 72°
I have assumed typical values for Ta and extension time. These may be modified as required by Pn and product size. I generally let Ta equal 10° less than the primer Tm. 15-30" is sufficient for products up to 1 kb in the AirCycler.
Electrophorese a 5 µL aliquot as before. Because the reamplification was done with a 25:1 ratio of Pn to Po, the smaller band should dominate, and may be the only one visible. If the contaminant band is not present or has a yield only a small fraction of that of the smaller band, then the remaining 95 µL of product can be cleaned up directly using a Wizard PCR purification. Otherwise, run the entire reaction on an agarose gel and purify DNA from the appropriate band.
The prep is now ready to be sequenced. Pn will prime from the known end, and Po from the unknown end.
Materials
1. 10x H, M or L-PCRB
500 mM Tris, pH 8.3
2.5 mg/mL BSA
MgCl2 to give 30, 20 or 10 mM, respectively
5% Ficoll
5 mM cresol red
2. T/TS
10.5 µL EDB
1 µL Taq Polymerase (Promega) @ 5 u/µL
1 µL TaqStart antibody (CloneTech) as delivered
3. EDB
2.5 mg/ml bovine serum albumin in 10 mM Tris, pH 8.3
References
1. S.R.J.A.M. Hermann, S. O'Neill, T.T. Tsao, R.M. Harding & J.L. Dale "Single primer amplification of flanking sequences" Biotechniques 29, 1176-1180 (2000)
2. E.C. Kofoid, C. Rappleye, I. Stojiljkovic & J. Roth, "The 17-gene ethanolamine (eut) operon of Salmonella typhimurium encodes five homologues of carboxysome shell proteins" J. Bacteriol. 181, 5317-5329 (1999)
Appendix -- Some Useful Primers
Primer common names are given first and should not be overinterpreted. Database names are in parentheses, followed by a comment. Sequences are written 5' to 3'. Each primer extends out of its associated cassette by the shortest possible route (that is, it emanates out of -- not into -- the cassette).
1. Tn10dTet core and derivatives
Po: TN10L (TP633) Binds just after tetA terminator.
ACCAACCATTTGTTAAATCAGTTTTTGTTGTGA
Po: TN10R (TP632) Binds just after tetR terminator.
CAGTGATCCATTGCTGTTGACAAAGGGAATC
Pn: Any appropriate IS10 primer below.
2. IS10 and most TN10 derivatives
Po: F1284 (TP89) 38 bp from end; will not work in TPOPs;
CAAGATGTGTATCTACCTTAAC
Po or Pn: IS10R2 (TP134) 27 bp from end; right side of TPOPs only;
CAAGATGTGTATCCACCTTAACTTAATGATTTT
Pn: IS10R4 (TP134) 4 bp from end; any Tn10 derivative, including TPOPs;
AACTTAATGATTTTGATCAAAATCATTAGGGGATTCA
3. MudJ and relatives
Po: R86 (TP251) 61 bp before left end.
GCAAGCCCCACCAAATCTAATCCCA
Pn: R54 (TP240) 36 bp before left end.
CCGAATAATCCAATGTCC
Po: F33 (TP81) 9 bp from end; extremely difficult PCR, because of large stem-loop; the first 5 cycles should each be preceded by a 5" hold at 94°C instead of the normal zero second holds; statistically, half the bands will be products extending into the mud element, not out.
GAAACGCTTTCGCGTTTTTCGTGCG
Pn: F20 (TP79) Exactly at end.
GTTTTTCGTGCGCCGCTTC
4. Tn5-derived chloramphenicol resistance cassette (found in Mud-cam and Tn10d-cam)
Po: CMR2 (TP699) 158 bp prior to gene facing upstream.
CTTCCCGGTATCAACAGGGACA
Pn: CMR1 (TP698) 214 bp prior to gene facing upstream.
GTCACAGGTATTTATTCGGCGCA
Po: CKO3 (TP45) 154 bp past gene facing downstream.
AGGGCAGGGTCGTTAAATAGC
Pn: CMR3 (TP700) 223 bp past gene facing downstream.
AGTGTGACCGTGTGCTTCTCAA