Methods for DNA isolation(DNA分离方法)
互联网
(adapted from Bruce A.Roe,Department of Chemistry and Biochemistry,The University of Oklahoma,Norman,Oklahoma 73019 broe@ou.edu)
A.Large scale double-stranded DNA isolation
The method used for the isolation of large scale cosmid and plasmid DNA is an unpublished modification (16)of an alkaline lysis procedure (17,18)followed by equilibrium μltracentrifugation in cesium chloride-ethidium bromide gradients (1).Briefly,cells containing the desired plasmid or cosmid are harvested by centrifugation,incubated in a lysozyme buffer,and treated with alkaline detergent.Detergent solubilized proteins and membranes are precipitated with sodium acetate,and the lysate is cleared first by filtration of precipitate through cheesecloth and then by centrifugation.The DNA -containing supernatant is transferred to a new tube,and the plasmid or cosmid DNA is precipitated by the addition of polyethylene glycol and collected by centrifugation.
The DNA pellet is resuspended in a buffer containing cesium chloride and ethidium bromide,which is loaded into polyallomer tubes and subjected to μltracentrifugation overnight.The ethidium bromide stained plasmid or cosmid DNA bands,equilibrated within the cesium chloride density gradient after μltracentrifugation,are visualized under long wave UV light and the lower band is removed with a 5 cc syringe.The intercalating ethidium bromide is separated from the DNA by loading the solution onto an equilibrated ion exchange column.The A260 containing fractions are pooled,diluted,and ethanol precipitated,and the final DNA pellet is resuspended in buffer and assayed by restriction digestion as detected on agarose gel electrophoresis.
During the course of this work several modifications to the above protocol were made.For example,initially cell growth times included three successive overnight incubations,beginning with the initial inoculation of 3 ml of antibiotic containing media with the plasmid or cosmid-containing bacterial colony,and then increasing the culture volume to 50 ml,and then to 4 l.However,it was observed that recombinant cosmid DNA isolated from cell cultures grown under these conditions,in contrast to recombinant plasmid DNA ,was contaminated with deleted cosmid DNA molecules.However,these deletions are avoided by performing each of the three successive incubations for eight hours instead of overnight,although a slight yield loss accompanied the reduced growth times.
Recently,a diatomaceous earth-based (19-22)method was used to isolate the plasmid or cosmid DNA from a cell lysate.The cell growth,lysis,and cleared lysate steps are performed as described above,but following DNA precipitation by polyethylene glycol,the DNA pellet is resuspended in RNase buffer and treated with RNase A and T1.Nuclease treatment is necessary to remove the RNA by digestion since RNA competes with the DNA for binding to the diatomaceous earth.After RNase treatment,the DNA containing supernatant is bound to diatomaceous earth in a chaotropic buffer of guanidine hydrochloride by incubation at room temperature.The DNA -associated diatomaceous earth then is collected by centrifugation,washed several times with ethanol buffer and acetone,dried,and then resuspended in buffer.The DNA is eluted during incubation at 65degC,and the DNA -containing supernatant is collected after centrifugation and separation of the diatomaceous earth particles.The DNA recovery is measured by taking absorbance readings at 260 nanometers.After concentration by ethanol precipitation,the DNA is assayed by restriction digestion.
Protocol
1.Pick a colony of bacteria harboring the plasmid or cosmid DNA of interest into a 12 X 75 mm Falcon tube containing 2 ml of LB media supplemented with the appropriate antibiotic (typically ampicillin at 100μg/ml)and incubate at 37deg C 8-10 hours with shaking at 250 rpm.Transfer the culture to an Ehrlenmeyer flask containing 50 ml of similar media,and incubate further for 8-10 hours.Transfer 12.5 ml of the culture to each of 4 liters of similar media,and incubate for an additional 8-10 hours.
2.Harvest the cells by centrifugation at 7000 rpm for 20 minutes in 500 ml bottles in the RC5-B using the GS3 rotor.Resuspend the cell pellets in old media and transfer to two bottles,centrifuge as before,and decant the media.The cell pellets can be frozen at -70degC at this point.
3.Resuspend the cell pellets in a total of 70 ml of GET/Lysozyme solution (35 ml for each bottle)by gently teasing the pellet with a spatula and incubate for 10 minutes at room temperature.(Note: Do not vortex the lysate at any time because this may shear the chromosomal DNA ).
4.Add a total of 140 ml of alkaline lysis solution (70 ml for each bottle),gently mix,and incubate for 5 minutes in an ice-water bath.
5.Add 105 ml of 3M NaOAc,pH 4.8 (52.5 ml for each bottle),cap tightly,gently mix by inverting the bottle a few times,and incubate in an ice-water bath for 30-60 minutes.
6.Clear the lysate of precipitated SDS,proteins,membranes,and chromosomal DNA by pouring through a double-layer of cheesecloth.Transfer the lysate into 250 ml centrifuge bottle,centrifuge at 10,000 rpm for 30 minutes at 4deg C in the RC5-B using the GSA rotor.
For cesium chloride-gradient purification:
7a.Pool the cleared supernatants into to a clean beaker,add one-fourth volume of 50% PEG/0.5 M NaCl,swirl to mix,and incubate in an ice-water bath for 1-2 hours.
8a.Collect the PEG-precipitated DNA by centrifugation in 250 ml bottles at 7000 rpm for 20 minutes at 4degC in the RC5-B using the GSA rotor.
9a.Dissolve the pellets in a combined total of 32 ml of 100:10 TE buffer,5 ml of 5 mg/ml ethidium bromide,and 37 g cesium chloride (Var Lac Oid Chemical Co.,Inc.)(final concentration of cesium chloride should be 1 g/ml).
10a.Transfer the sample into 35 ml polyallomer centrifuge tubes,remove air bubbles,seal with rubber stoppers,and crimp properly.
11a.Centrifuge at 60,000 rpm to 16-20 hours at 15-20degC in the Sorvall OTD-75B μltracentrifuge (DuPont)using the T-865 rotor.
12a.Visualize the ethidium bromide stained DNA under long-wave UV light,and remove the lower DNA band using a 5 cc syringe and a 25 gauge needle.(It may be helpful first to remove and discard the upper band).
13a.To remove the ethidium bromide,load the DNA sample onto an equilibrated 1.5 ml Dowex column,and collect 0.5 ml fractions.Equilibrate the Dowex AG resin (BioRad)by successive centrifugation,resuspension,and decanting with 1M NaOH,water,and then 1M Tris-HCl,pH 7.6 until the Dowex solution has a pH of 7.6.
14a.Pool fractions with an A260 of 1.00 or greater into 35 ml Corex glass tubes,add one volume of ddH2O,and ethanol precipitate by adding 2.5 volumes of cold 95% ethanol.Incubate at least 2 hours at -20degC,centrifuge at 10,000 rpm for 45 minutes in the RC5-B using the SS-34 rotor.Gently decant the supernatant,add 80% ethanol,centrifuge as before,decant,and dry the DNA pellet in a vacuum oven.
15a.Resuspend the DNA in 10:0.1 TE buffer.
For diatomaceous earth-based purification:
7b.Pool the supernatants from step 6 into 500 ml bottles and add DNase-free RNase A and RNase T1 such that the final concentration of RNase A is 40μg/ml and RNase T1 is 40 U/ml.Incubate in a 37degC water bath for 30 minutes.
8b.Add an equal volume of isopropanol and precipitate at room temperature for 5 minutes.Centrifuge at 9,000 rpm for 30 minutes in the RC5-B using the GS3 rotor.Decant the supernatant and drain the DNA pellet.
9b.Resuspend each DNA pellet in 20 ml 10:1 TE buffer,and add 40 ml of de-fined diatomaceous earth in guanidine-HCl (100 mg/ml)to each bottle.Allow the DNA to bind at room temperature for 5 minutes with occasional mixing.Centrifuge at 9,000 for 10 minutes in the RC5-B using the GS3 rotor.
10b.Decant the supernatant,resuspend each pellet in 40 ml of diatomaceous earth-wash buffer,and centrifuge as above.
11b.Decant the supernatant,resuspend each pellet in 40 ml of acetone,and centrifuge as above.
12b.Decant the supernatant and dry the pellet in a vacuum oven.
13b.Resuspend the pellet in 20 ml of 10:1 TE buffer,and elute the bound DNA by incubation at 65degC for 10 minutes with intermittent mixing.
14b.Remove the diatomaceous earth by centrifugation at 9,000 rpm for 10 minutes in the RC5-B using the GS3 rotor.Repeat if necessary.
15b.Combine the DNA -containing supernatants and precipitate the DNA in 35 ml Corex glass tubes adding 2.5 volumes of cold 95% ethanol/acetate.
16b.Resuspend the dried DNA pellet in 2 ml of 10:0.1 TE buffer and assay for concentration by absorbance readings at 260 nm or by agarose gel electrophoresis.
B.Midiprep double-stranded DNA isolation
A midi-prep double-stranded DNA isolation has been developed to generate a sufficient amount of template DNA for several Sequenase[TM] catalyzed fluorescent terminator reactions.Here,one bacterial colony which harbored the plasmid of interest is picked into 3 ml of liquid media containing ampicillin and incubated in a 37degC shaker for 8-10 hours.At this time,the culture is transferred into 50 ml of ampicillin-containing media and incubated further for 10-12 hours.After harvesting the cells by centrifugation,a diatomaceous earth-based alkaline-lysis purification method (19-22)is performed,similar to that discussed above for large scale DNA isolation.The purified DNA is crudely assayed for concentration and purity by agarose gel electrophoresis against known standards.The approximate yield of double-stranded DNA using this method is 1μg of DNA per ml of cell culture.For a 50 ml cell culture,about 50μg of DNA are recovered,and 5μg are used typically in a Sequenase[TM] terminator reaction.
Note: This procedure is the method of choice for isolating double stranded plasmid-based templates for the Sequenase Dye-Labeled Terminator Sequencing Reactions.
Protocol
1.Pick a colony of bacteria harboring the plasmid DNA of interest into a 12 X 75 mm Falcon tube containing 3 ml of 2xTY media supplemented with the appropriate antibiotic (typically ampicillin at 100μg/ml)and incubate at 37deg C 8-10 hours with shaking at 250 rpm.Transfer the culture to an Ehrlenmeyer flask containing 50 ml of similar media,and incubate further for 11-14 hours.
2.Harvest the cells by centrifugation at 3000 rpm for 5 minutes in 50 ml conical tubes in the Beckman GPR tabletop centrifuge and decant the supernatant.The cell pellets can be frozen at -70degC at this point.
3.Resuspend the cell pellets in 2 ml of GET/Lysozyme solution,add 4 ml of alkaline lysis solution,gently mix,and incubate for 5 minutes in an ice-water bath.
4.Add 4 ml of 3M NaOAc,pH 4.8,gently mix by swirling,and incubate in an ice-water bath for 30-60 minutes.
5.Clear the lysate of precipitated SDS,proteins,membranes,and chromosomal DNA by pouring through a double-layer of cheesecloth into a new 50 ml conical tube.Centrifuge at 3,000 rpm for 20 minutes at 4degC in the Beckman GPR tabletop centrifuge.
6.Decant the supernatant to a 50 ml polypropylene centrifuge tube,add 20μl of a 20 mg/ml DNase-free RNase A and incubate in a 37degC water bath for 30 minutes.
7.Add 7 ml (equal volume)of de-fined diatomaceous earth in guanidine-HCl (20 mg/ml)and allow the DNA to bind at room temperature for 5 minutes with occasional mixing.Centrifuge at 3,000 for 5 minutes in the Beckman GPR tabletop centrifuge.
8.Decant the supernatant,resuspend in 7 ml of diatomaceous earth-wash buffer,and centrifuge as above.
11.Resuspend the pellet in 0.6 ml of 10:1 TE buffer,and elute the bound DNA by incubation at 65degC for 10 minutes with intermittent mixing.
12.Remove the diatomaceous earth by centrifugation at 3,000 rpm for 5 minutes in the in the Beckman GPR tabletop centrifuge.
13.Transfer the supernatant to a 1.5 ml microcentrifuge tube and centrifuge at 12,000 rpm for 5 minutes in a microcentrifuge at room temperature.Transfer the supernatant to a new 1.5 ml microcentrifuge tube and ethanol precipitate.
14.Resuspend the dried DNA pellet in 40μl of 10:0.1 TE buffer and assay for concentration by agarose gel electrophoresis.
C.Miniprep double-stranded DNA isolation
The standard method for the miniprep isolation of plasmid DNA includes the same general strategy as the large scale isolation.However,smaller aliquots of antibiotic containing liquid media inoculated with plasmid-containing cell colonies are incubated in a 37degC shaker for 12-16 hours.After collecting the plasmid containing cells by centrifugation,the cell pellet is resuspended in a hypotonic sucrose buffer.The cells are successively incubated with an RNase-lysis buffer,alkaline detergent,and sodium acetate.The lysate is cleared of precipitated proteins and membranes by centrifugation,and the plasmid DNA is recovered from the supernatant by isopropanol precipitation.The DNA is crudely checked for concentration and purity using agarose gel electrophoresis against known standards.A typical yield for this method of DNA isolation is 10-15μg of plasmid DNA from a 6 ml starting culture.
Since highly supercoiled DNA is desired for double-stranded DNA sequencing,a modification of this method employing diatomaceous earth (19-22)sometimes is used for isolation of double-stranded templates for DNA sequencing with fluorescent primers.After removal of the precipitated proteins and membranes,the plasmid containing supernatant is incubated with diatomaceous earth and guanidine hydrochloride and this mixture is added into one of the twenty-four wells in the BioRad Gene Prep Manifold.The supernatant is removed by vacuum filtration over a nitrocellulose filter.The DNA -associated diatomaceous earth is washed to remove the guanidine hydrochloride with an ethanol buffer,and then dried by filtration.Elution buffer is added to the wells,and the DNA -containing solution then is separated from the diatomaceous earth particle by filtration into a collection tube.The collected DNA is concentrated by ethanol precipitation and crudely assayed for concentration and purity by agarose gel electrophoresis against known standards.The approximate yield of double-stranded DNA is 3-5μg of DNA from 6 ml of starting culture.
Note: This is a typical mini-prep until step 7,where in step 7a you would precipitate the template and use it for Taq Cycle Sequencing with the Dye-Labeled Primers,or in step 7b proceed with the diatomaceous earth purification for Taq Dye-Labeled Terminator Cycle Sequencing Reactions.For Sequenase Dye-Labeled Terminator Sequencing Reactions use the Midi-prep procedure detailed above.
Protocol
1.Pick a colony of bacteria harboring the plasmid DNA of interest into a 17 X 100 mm Falcon tube containing 6 ml of TB media supplemented with the appropriate antibiotic (typically ampicillin at 100μg/ml)and incubate at 37deg C 16-18 hours with shaking at 250 rpm.
2.Harvest the cells by centrifugation at 3000 rpm for 5 minutes in the Beckman GPR tabletop centrifuge and decant the supernatant.The cell pellets can be frozen at -70degC at this point.
3.Resuspend the cell pellets in 0.2 ml of TE-RNase solution (50:10 TE buffer containing 40μg/ml RNase A; some also add RNase T1 to a final concentration of 10 U/μl)by gentle vortexing,add 0.2 ml of alkaline lysis solution,gently mix,and incubate for 15 minutes at room temperature.
4.Add 0.2 ml of 3M NaOAc,pH 4.8,gently mix by swirling,transfer to 1.5 ml microcentrifuge tubes,and incubate in an ice-water bath for 15 minutes.
5.Clear the lysate of precipitated SDS,proteins,membranes,and chromosomal DNA by centrifugation at 12,000 rpm for 15 minutes in a microcentrifuge at 4deg C.
6.Transfer the supernatant to a fresh 1.5 ml microcentrifuge tube,incubate in an ice-water bath for 15 minutes,centrifuge as above for an additional 15 minutes and transfer the supernatant to a clean 1.5 ml tube.
For standard alkaline lysis purification:
7a.Precipitate the DNA by adding 1 ml of 95% ethanol,and resuspend the dried DNA pellet in 100-200μl 10:0.1 TE buffer.Electrophorese an aliquot of the DNA sample on a 0.7% agarose gel to crudely determine the concentration and purity.
For diatomaceous earth-base purification:
7b.Add 1 ml of de-fined diatomaceous earth in guanidine-HCl (20 mg/ml)and allow the DNA to bind at room temperature for 5 minutes with occasional mixing.Meanwhile soak the Prep-A-Gene nitrocellulose membrane in isopropanol for at least 3 minutes,and assemble the Prep-A-Gene manifold as described in the manual.
8b.Turn on the vacuum pump and adjust the vacuumlevel to 8 in.Hg,let the membrane dry for 1 minute,and then release the vacuum.
9b.Pour the well mixed samples into the wells of the Prep-A-Gene manifold and filter through at 8 in.Hg until all the liquid is filtered through.
10b.Wash the samples four times with 250μl of diatomaceous earth-wash buffer,using a repeat pipette,allowing all of the liquid to filter through between washes.
11b.Reduce the vacuum to 5 in.Hg before turning the vacuum off at the stopcock.Without unscrewing the black clamps,release the white clamps and place the collection rack with clean 1.5 ml screw-capped tubes into the manifold.Clamp the manifold with the white clamps,and apply 300μl of 10:1 TE buffer heated to 65degC and pull the eluted DNA through at 5 in.Hg.After the liquid has filtered through,raise the vacuum to 10-12 in.Hg,and let the membrane dry for 1 minute.
12b.Turn off the vacuum at the stopcock and remove the collection rack containing the tubes.Ethanol precipitate the DNA and resuspend the dried DNA pellet in 30μl of 10:0.1 TE buffer.
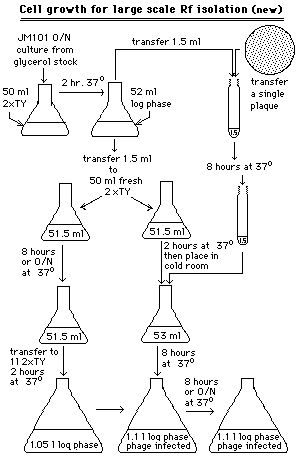
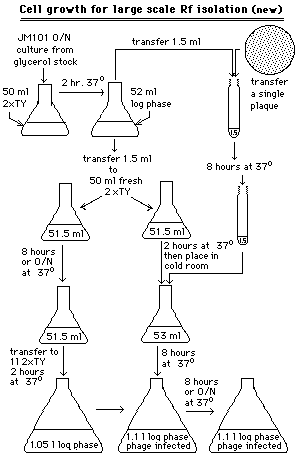
D.Large scale M13RF isolation (9)
Double-stranded M13RF is isolated for use in M13 SmaI cut,dephosphorylated vector preparation,described below.The growth conditions of M13-infected bacterial cells (see Figure 1)appears convoluted,but result in a maximal amount of M13 RF molecules per cell.After the M13RF containing bacterial cells are harvested by centrifugation,the double-stranded molecules are isolated using the cesium chloride method for large scale plasmid isolation,as described above.This briefly entailed alkaline cell lysis,sodium acetate precipitation of detergent solubilized proteins and membranes,polyethylene glycol DNA precipitation,and extraction of ethidium bromide stained DNA from a cesium chloride gradient after μltracentrifugation.After removal of the ethidium bromide on an ion-exchange column,the DNA containing fractions are detected by A260 measurement and pooled,and the DNA is concentrated by ethanol precipitation and assayed by restriction enzyme digestion and agarose gel electrophoresis.
Protocol
1.Prepare an early log phase culture of JM101 by inoculating an Ehrlenmeyer flask containing 50 ml of 2xTY with a glycerol stock of JM101 and pre-incubating for 1 hour in a 37degC water bath,with no shaking.Pick a plaque representing the desired M13 clone into four 1.5 ml aliquot of early log phase JM101,and incubate according to the procedure displayed in Figure 1 to result in 4 liters of M13-infected bacteria.
2.Harvest the cells by centrifugation at 7000 rpm for 20 minutes in 500 ml bottles in the RC5-B using the GS3 rotor.Resuspend the cell pellets in fresh 2xTY media to remove contaminating extracellular phage and transfer to two bottles,centrifuge as before,and decant the media.The cell pellets can be frozen at -70degC at this point.
3.Resuspend the cell pellets in a total of 120 ml (30 ml for each bottle)of 1X STB buffer by gently teasing the pellet with a spatula.Add a total 24 ml of lysozyme solution (6 ml for each bottle),gently mix,and incubate for 5 minutes in an ice-water bath.
4.Add 48 ml of 50:2:10 TTE buffer (12 ml for each bottle)and 2 ml of RNase A (10 mg/ml)(0.5 ml for each bottle),gently mix,and incubate in an ice-water bath for 5 minutes.
5.Clear the lysate of precipitated SDS,proteins,membranes,and chromosomal DNA by pouring through a double-layer of cheesecloth.Transfer the lysate into 250 ml centrifuge bottle,centrifuge at 10,000 rpm for 30 minutes at 4deg C in the RC5-B using the GSA rotor.
6.Add 6 ml of 5 mg/ml ethidium bromide,and cesium chloride such that the final concentration of cesium chloride is 1 g/ml.
7.Transfer the sample into 35 ml polyallomer centrifuge tubes and top off with a 1:1 solution of 100:10 TE buffer and cesium chloride,remove air bubbles,seal with rubber stoppers,and crimp properly.
8.Centrifuge at 60,000 rpm to 16-20 hours at 15-20degC in the Sorvall OTD-75B μltracentrifuge using the T-865 rotor.
9.Visualize the ethidium bromide stained DNA under long-wave UV light,and remove the lower DNA band using a 5 cc syringe and a 25 gauge needle.(It may be helpful to remove and discard the upper band first).
10.To remove the ethidium bromide,load the DNA sample onto an 1.5 ml Dowex AG (BioRad)column,equilibrated as before,and collect 0.5 ml fractions.
11.Pool fractions with an A260 of 1.00 or greater into 35 ml Corex glass tubes,add one volume of ddH2O,and ethanol precipitate by adding 2.5 volumes of cold 95% ethanol.Incubate at least 2 hours at -20degC,centrifuge at 10,000 rpm for 45 minutes in the RC5-B using the SS-34 rotor.Gently decant the supernatant,add 80% ethanol,centrifuge as before,decant,and dry the DNA pellet in a vacuum oven.
12.Resuspend the DNA in 10:0.1 TE buffer.[Image]
E.Single-stranded M13 DNA isolation using phenol
This isolation procedure (23)is the method of choice for preparation of M13-based templates to be used in Sequenase[TM] catalyzed dye-terminator reactions.A pre-incubated early log phase JM101 culture is prepared by transferring a thawed glycerol stock into 50 ml of liquid media and incubating for 1 hour at 37degC with no shaking.M13 plaques are picked with a sterile toothpick and placed into 1.5 ml aliquots of the early log phase JM101 culture,which are incubated in a 37deg C shaker for 4-6 hours.After incubation,the bacterial cells are pelleted by centrifugation and the viral containing supernatant is transferred to a clean tube.The phage particle are precipitated with PEG,collected by centrifugation,and the pellet is resuspended in buffer.The phage protein coat is denatured and removed by one phenol and two ether extractions.After ethanol precipitation,the dried DNA pellet is resuspended in buffer,and the concentration and purity crudely are assessed by agarose gel electrophoresis against known standards.
Protocol
1.Prepare an early log phase culture of JM101,as above,and pick M13-based plaques with sterile toothpicks into 12 X 75 mm Falcon tubes containing 1.5 ml aliquots of the cells.Incubate for 4-6 hours at 37degC with shaking at 250 rpm.
2.Transfer the culture to 1.5 ml microcentrifuge tubes and centrifuge for 15 minutes at 12,000 rpm at 4degC.
3.Pipette the top 1 ml of supernatant to a fresh 1.5 ml microcentrifuge tube containing 0.2 ml 20% PEG/2.5 M NaCl to precipitate the phage particles.Mix by inverting several times and incubate for 15-30 minutes at room temperature.
4.Centrifuge for 15 minutes at 12,000 rpm at 4degC to collect the precipitated phage.Decant the supernatant and remove residual PEG supernatant by suctioning twice.
5.Resuspend the pellet in 100μl of 10 mM Tris-HCl,pH 7.6 by vortexing,and add 50μl of TE-saturated phenol.
6.Extract the DNA with phenol and twice with ether,as discussed above,and then ethanol precipitate.
7.Resuspend the dried DNA in 6μl of 10:0.1 TE for use in single-stranded Sequenase[TM] catalyzed dye-terminator sequencing reactions.
F.Biomek-automated modified-Eperon isolation procedure for single-stranded M13 DNA
This semi-automated method is a modification of a previously reported procedure (24,25),and allowed the simultaneous isolation