Avoiding DNA Contamination in RT-PCR
互联网
Avoiding DNA Contamination in RT-PCR
A frequent cause of concern among investigators performing quantitative RT-PCR is inaccurate data due to DNA contamination in RNA preparations. Although DNA contamination is easily detected by performing a "no-RT" control, there is no easy remedy. In this technical bulletin, we present data showing levels of DNA contamination in RNA generated by different procedures, and suggest several precautionary measures that can be implemented to reduce the impact of this persistent problem.
RT-PCR and Genomic Contamination
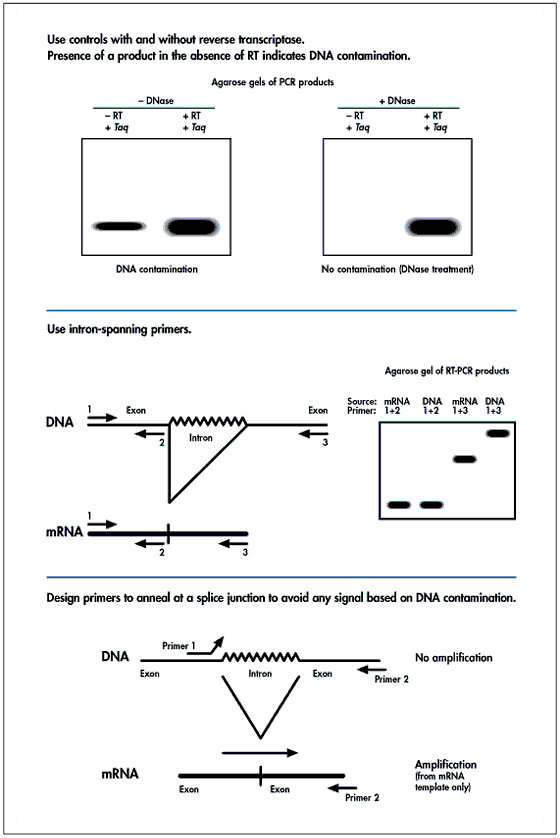
RT-PCR is an increasingly popular method for the quantitative analysis of gene expression. With this popularity comes a heightened awareness that most techniques used for total RNA isolation yield RNA with significant amounts of genomic DNA contamination. PCR cannot discriminate between cDNA targets synthesized by reverse transcription and genomic DNA contamination. At Ambion, we can routinely perform PCR from residual genomic DNA present in total RNA samples isolated by most commonly used techniques. To illustrate this problem, we performed RT-PCR on mouse liver RNA isolated by a multi-step guanidinium thiocyanate/acid phenol:chloroform extraction (ToTALLY RNA™), a one-step extraction (Tri Reagent), a filter-binding based extraction (RNAqueous™), by centrifugation through a CsCl cushion, and by two rounds of oligo d(T) selection using Ambion's Poly(A)Pure™ Kit (see Figure 1a ). Regardless of whether reverse transcriptase was added in the reverse transcription step, gene specific product is synthesized in most samples. Among the total RNA samples, the amount of DNA contamination is lowest in the CsCl-pelleted RNA. No signal is apparent in the oligo d(T)-selected sample. The PCR products in the "no-RT" samples are the result of amplification from trace amounts of genomic contamination.
<center> <table> <tbody> <tr> <td> <div> <a name="1"> <font> </font> </a></div><a name="1"> </a><a name="1"> </a></td> </tr> <tr> <td> <p> <font><font><span>Figure 1.</span> <span>DNA Contamination in RNA Isolated by Five Different Methods.</span> <span>Mouse liver total RNA was isolated according to protocol by five different methods. 0.5 µg RNA was used in RT-PCR reactions with Ambion's RETROscript?Kit. PCR reactions were performed with 5 µg RNA. 10 µl of each reaction was electrophoresed on a 2% agarose gel and stained with EtBr. </span> </font> </font></p> <div> <table> <tbody> <tr> <td> <p class="figure-title"> <font>Lane </font></p> </td> <td> <p class="figure-title"> <font>RNA Isolation Method </font></p> </td> </tr> <tr> <td> <p> <font>1 </font></p> </td> <td> <p> <font>One Step RNA Isolation (Tri Reagent) </font></p> </td> </tr> <tr> <td> <p> <font>2 </font></p> </td> <td> <p> <font>Glass Binding Method (Ambion's RNAqueous™ Kit) </font></p> </td> </tr> <tr> <td> <p> <font>3 </font></p> </td> <td> <p> <font>Acid Phenol Chloroform Method (Ambion's ToTALLY RNA™ Kit) </font></p> </td> </tr> <tr> <td> <p> <font>4 </font></p> </td> <td> <p> <font>CsCl cushion </font></p> </td> </tr> <tr> <td> <p> <font>5 </font></p> </td> <td> <p> <font>Oligo dT Selection (Ambion's Poly(A)Pure™ Kit) </font></p> </td> </tr> <tr> <td> <p> <font>6 </font></p> </td> <td> <font>H<sub>2</sub> O Control</font></td> </tr> </tbody> </table> </div> </td> </tr> </tbody> </table> </center>
Differential Enrichment by Oligo d(T) Selection
Although two rounds of oligo d(T) selection are sufficient to remove genomic DNA contamination, there are two drawbacks to using this technique to control for DNA contamination. First, oligo d(T) chromatography is expensive and labor intensive for routine analysis. Secondly, a potentially serious problem not usually addressed is that relative amounts of individual transcripts can change with oligo d(T) chromatography, probably as a result of differential polyadenylation between tissues or in response to stimuli. At Ambion, we have found that oligo d(T) selection can even change the apparent abundance of transcripts from genes that are thought to have invariant expression. For example, when we compare the relative enrichment of cyclophilin and GAPDH transcripts by Northern blot analysis of total versus oligo d(T) selected mouse RNA, we see an obvious change in the apparent abundance of these two transcripts. As shown in Figure 2 , oligo d(T) selection enriches GAPDH and cyclophilin 17X and 22X, respectively, from kidney RNA, but 21X and 28X from thymus RNA. The source of this variation is unclear, but the implications for quantitation from oligo d(T) selected RNA are impossible to ignore.
<center> <table> <tbody> <tr> <td> <div> <a name="2"> <font> </font> </a></div><a name="2"> </a><a name="2"> </a></td> </tr> <tr> <td> <font><font><span>Figure 2. </span> <span>Differential Enrichment of Specific mRNAs by Oligo dT Chromatography. </span> <span>A Northern blot containing total RNA (1 µg) and twice oligo d(T) selected RNA (50 ng) from mouse thymus and kidney was hybridized simultaneously with GAPDH and cyclophilin RNA probes. Hybridization signals were quantitated with a Bio-Rad Molecular Imager. </span> </font> </font></td> </tr> </tbody> </table> </center> <center> </center>
<center> </center>
Primer Design
Primers for quantitative experiments are typically designed to amplify a target between 150 and 600 base pairs. Targets smaller than 200 bp are difficult to resolve on agarose gels, and larger targets place a greater burden on the investigator to optimize PCR conditions. The critical aspect for RT-PCR primer choice with respect to minimizing the problems associated with DNA contamination is to design primers that span at least one intron of the genomic sequence. This will result in a PCR product from genomic contamination that will be larger in size than the product generated from the cDNA. In fact, primers can be designed to span a sufficiently large genomic fragment such that amplification from contaminating DNA may be not be possible. For genes in which the genomic sequence is published, the positions of the splice junctions can be found by retrieving the sequence from the Genbank database at http://www.ncbi.nlm.nih.gov/Genbank/index.html . If the intron - exon structure is unknown, primers can be synthesized in different regions of the cDNA sequence and tried in combinations on both cDNA and genomic DNA. It should be possible to choose a primer combination that yields either no product (additional intron sequence produces too long a target for efficient PCR) or an easily distinguishable product when amplifying from genomic DNA. An additional wrinkle is that pseudogenes exist in the mammalian genome for many genes, including the most commonly used internal controls (ß-actin, GAPDH, cyclophilin). These sequences, arising from integration of a reverse transcription product into the genome, do not have introns. Thus, the size of a PCR product amplified from a pseudogene may be identical to that produced from a cDNA copy. The only way to identify these products is to perform a "no-RT" control as shown in Figure 3 . The true product from RNA is 361 base pairs. The "no-RT" control yields both a fragment identical in size to the expected RT-PCR product from the RNA transcript (from a pseudogene), and a 1.2 kb fragment from the legitimate genomic locus. If it is absolutely essential to avoid amplification from these sequences, an amplified fragment from a pseudogene may be sequenced, and primers designed to regions where the sequence varies from the legitimate copy of the gene. As little as a one or two nucleotide difference at the 3' end of a primer binding site can completely inhibit amplification from the pseudogene.
<center> <table> <tbody> <tr> <td> <div> <font><font><font><font><a name="3"> <font> </font> </a></font></font></font></font></div><a name="3"> <font><font><font><font></font></font></font></font></a><font><font><font><a name="3"> </a></font></font></font></td> </tr> <tr> <td> <font><font><font><font><font><font><span>Figure 3</span> . <span>DNA Contamination in RNA.</span> <span>Mouse liver total RNA was isolated according to protocol. RT-PCR reactions were performed using Ambion's RETROscript?Kit and 0.5 µg RNA. PCR reactions were performed with 5 µg RNA. 10 µl of each reaction was electrophoresed on a 2% agarose gel and stained with EtBr.</span> </font> </font> </font></font></font></font></td> </tr> </tbody> </table> <p> <font><font><font><font><font> </font></font></font></font></font></p> <p> <font><font><font><font><font><span><font>DNase I Treatment</font> </span> </font></font></font></font></font></p> <p> <font><font><font><font><font><span><font>In a recent informal survey of RT-PCR users, we found that the field is evenly divided by those users who believe that DNase I treatment solves the problem of genomic DNA contamination and those who feel that DNase I treatment is an unacceptable solution. Detractors claim that DNase I treatment and the subsequent inactivation steps compromise the performance of their RT-PCR reactions to an unacceptable degree. Much of the problem these users experience may be traced to the extreme temperatures used to inactivate the DNase I prior to reverse transcription. Huang, et al. (<i>Biotechniques</i> , (1996) 20:(6)1012-1020) report complete inactivation of DNase I by heat denaturation at 75°C for 5 minutes. Lower inactivation temperatures do not completely inactivate DNase I, while higher temperatures appear to damage the RNA template. DNase I treatment followed by heat inactivation is a simple enough technique for routine use in systems in which genomic DNA contamination is a problem. The use of high quality, RNase-free DNase is crucial. Two additional conventional methods of reducing contaminating genomic DNA from total RNA preparations are acid phenol extraction, which partitions DNA into the organic phase, and LiCl precipitation, which selectively precipitates RNA from solution (protein and DNA remain in the supernatant). A description of these techniques can be found in Ambion's Technical Bulletins #</font> <font>158</font> <font> and #</font> <font>160</font> <font>. These techniques can be used after DNase I treatment to inactivate the enzyme and precipitate the RNA prior to reverse transcription. Finally, it should be noted that DNase I treatment neither relieves the investigator of the burden of sensible primer design, nor of the necessity to perform the appropriate "no-RT" controls. </font> </span> </font></font></font></font></font></p> </center>
In addition to the above techniques, researchers now have a new and convenient option for removal of DNA and DNase I from RNA samples. DNA-free ™ DNase Treatment and Removal Reagents are designed for the removal of contaminating DNA from RNA samples and for the removal of DNase after treatment. As described above, DNase is typically inactivated by heat treatment, and can also be removed from treated preps by phenol extraction. Heat inactivation can present problems, however, as the temperature at which DNase is inactivated also catalyzes RNase-independent RNA strand scission in the presence of divalent cations. Phenol extraction is also avoided by researchers who do not want to work with phenol, or who worry about sample loss.
DNA-free avoids both methods of DNase I inactivation by supplying a novel DNase Removal reagent that effectively removes DNase and divalent cations from the reaction mixture. The DNase/cation removal step takes a mere three-minute incubation. No organic extraction, EDTA addition or heat inactivation is required.
The DNA-free DNase Treatment and Removal Reagents are provided with RNase-free DNase I, an optimized 10X Reaction Buffer, and the DNase Removal Reagent. The DNA-free Reagents are now also part of the RNAqueous™-4PCR Kit, combining the features and benefits of RNAqueous™ with those of DNA-free.
In addition to DNA-free , Ambion offers many quality products to facilitate successful RT-PCR experiments. These include RNase-free pipette tips and PCR tubes, RNase free DNase I, ToTALLY RNA, RNAqueous, and Poly(A)Pure RNA Isolation Kits, RETROscript First Strand cDNA Synthesis Kit, and SuperTaq?thermostable DNA polymerase. All of Ambion's products designed for use with RNA undergo rigorous quality testing and are certified RNase-free.
The Polymerase Chain Reaction (PCR) is covered by patents owned by Hoffman-LaRoche. Use of the PCR process requires a license. A license for research may be obtained by purchase and use of authorized reagents and DNA thermal cyclers.
SuperTaq™ is made by Enzyme Technologies Limited and sold under licensing arrangements with F. Hoffmann-La Roche Ltd., Roche Molecular Systems, Inc. and the Perkin-Elmer Corporation. Ambion is a distributor of Enzyme Technologies Limited.
Purchase of SuperTaq is accompanied by a limited license for its use in the Polymerase Chain Reaction (PCR) and RT-PCR process for research in conjunction with a thermal cycler, the use of which in the automated performance of the PCR and RT-PCR process is covered by the up-front license fee, either by payment to Perkin-Elmer, or as purchased, i.e., an authorized thermal cyler.
Super Taq is not available for sale directly from Ambion in the United Kingdom, France, BeNeLux, Denmark, Sweden, Italy, Austria, Switzerland, Singapore, and Taiwan. Contact Enzyme Technologies LTD, Unit 4, 61 Ditton Walk, Cambridge CB5 8QD, U.K. (phone 44-1223-412-583) for more information.