反转录过程中需要考虑哪些因素?
丁香实验
为了研究 RNA 的功能,通常需要通过反转录过程将 RNA 转化为更稳定的互补 DNA(cDNA)。之后 cDNA 可通过分子克隆、PCR 和测序等技术做进一步的研究。因此,反转录过程是许多 RNA 实验研究流程的关键步骤。
为了获得更为准确的研究结果,本期总结了反转录过程中需要考虑的一些因素,以期能够抛砖引玉,给你的实验研究带来些许帮助。
本期内容
- RNA 模板制备
- 基因组 DNA 的去除
- 反转录酶选择
- 引物选择
- 主要反应组分
- 反应温度和反应时间
- 第一链和第二链 cDNA 合成
注:本期内容较多,阅读完此文需要大概 8 分钟。
一、RNA 模板制备
RNA 是反转录的模板。总 RNA 通常用于 RT-(q)PCR 等下游应用的 cDNA 合成,而特定类型的 RNA(如信使 RNA(mRNA)、miRNA 等小 RNA)可通过富集而用于某些实验应用,如 cDNA 文库构建和 miRNA 图谱分析。
保持 RNA 的完整性至关重要,在提取、处理、储存和实验过程中均需要采取特殊的保护措施。防止RNA降解的最佳方法包括佩戴手套、使用具有气溶胶屏障的移液器移液、使用无核酸酶的实验室器具和试剂以及对实验区域进行去污处理。
根据来源材料(如血液、组织、细胞、植物)的类型和实验目标,有多种 RNA 分离和纯化方法可供选择。分离纯化过程的主要目标是稳定 RNA 分子、抑制 RNA 酶,并通过适当的储存和提取方法最大程度提高产量。最佳的纯化方法可以去除干扰酶活性的内源性化合物,如植物组织中的复合多糖和腐殖酸,以及反转录酶的常见抑制剂,如盐、金属离子、乙醇和苯酚。纯化的 RNA 应保存在 -80°C,尽量减少反复冻融。
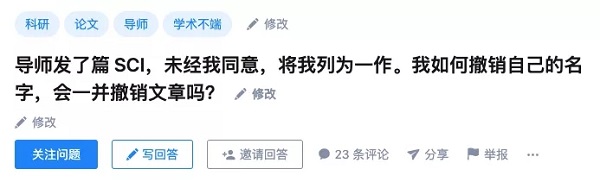
纯化后评估 RNA 质量和数量的方法有许多种。常用的方法是利用紫外光谱法检测特定波长的吸光度。RNA 质量可通过利用 Beer-Lambert 定律测定 260 nm 处的吸光度而确定;不同波长的吸光度比值可以确定是否存在特定污染物(表 1)。请注意,紫外吸光度不是 RNA 特有的,所有核酸都可以吸收相近波长的紫外线。对于需要更高 RNA 特异和更灵敏分析的 RNA, 可以考虑使用含染料的荧光试剂,这种试剂只在特异性的与目标分子结合时才会释放荧光信号。
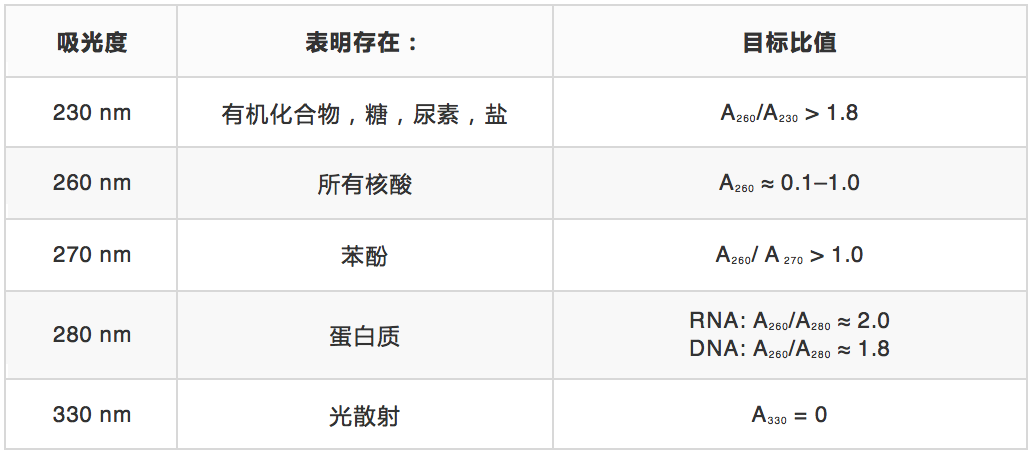 表 1. RNA 分析的紫外检测指南
表 1. RNA 分析的紫外检测指南
以 28S 和 18S 核糖体 RNA(rRNA)的比值可评估 RNA 的完整性。将总 RNA 变性后进行凝胶电泳,可对其进行定性评估。然后,评估 28S rRNA 与 18S rRNA 的强度比,比值为 2:1 代表完整的 RNA(图1A)。Agilent Technologies 开发的一种方法结合了微流体学和专利算法,可定量评估 RNA 的完整性。该方法可产生数字读数,称为 RNA 完整性分数或 RIN 值,数值介于 8 至 10 之间代表高质量的 RNA [1,2](图1B)。
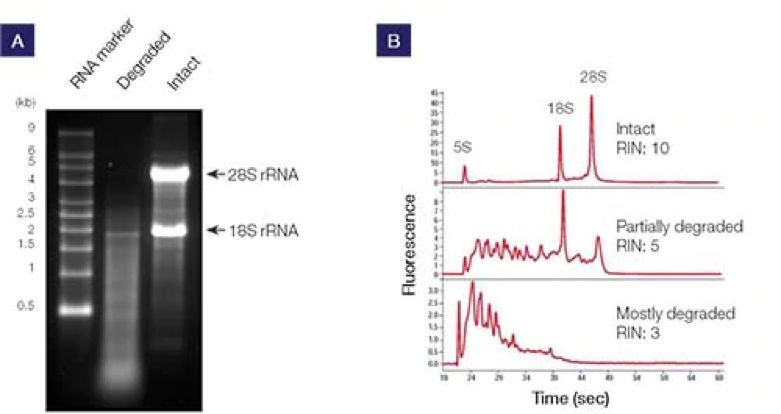 图1. 利用(A)凝胶电泳和(B)微流体学方法进行 RNA 完整性分析。
图1. 利用(A)凝胶电泳和(B)微流体学方法进行 RNA 完整性分析。
二、基因组 DNA 的去除
某些情况下,痕量基因组 DNA(gDNA)可能与 RNA 共同被纯化。污染 gDNA 可能会干扰反转录结果,导致 RT-qPCR 等高灵敏度实验出现假阳性、高背景或检测率降低。
为去除 gDNA,通常在 RNA 分离过程中加入 DNase I。在进行 RT-(q)PCR 之前,必须完全去除 DNase I,因为任何残留的酶都会使单链 DNA(如引物和 cDNA)降解。通常,DNase I 失活(如,EDTA 和加热处理)或酶去除步骤会导致 RNA 降解或样品损失。
作为 DNase I 的替代品,可使用双链特异性 DNase 去除污染 gDNA,而不影响 RNA 或单链 DNA。它们的热分解性质使其在相对温和的温度(如 55℃)下即可失活,同时没有负面影响。在反转录反应之前,只需将这种双链特异性 DNase 与 RNA 在 37℃ 下孵育 2 分钟,从而简化实验流程(图 2)。
 图 2. gDNA去除步骤:DNase I 与 Invitrogen™ ezDNase™ 酶。与 DNase I 相比,ezDNase 酶具有更短的工作流程、简单的处理步骤和较少的 RNA 损伤。可选择在反转录前使 ezDNase 酶失活,因为酶不会切割引物、ssRNA 或 cDNA:RNA 复合物。
图 2. gDNA去除步骤:DNase I 与 Invitrogen™ ezDNase™ 酶。与 DNase I 相比,ezDNase 酶具有更短的工作流程、简单的处理步骤和较少的 RNA 损伤。可选择在反转录前使 ezDNase 酶失活,因为酶不会切割引物、ssRNA 或 cDNA:RNA 复合物。
三、反转录酶选择
反转录酶以 RNA 为模板合成 cDNA,但这一类反转录酶可能具有不同的功能活性和性质。它们的性质会影响其反转录长链 RNA、高 GC 含量 RNA、具有复杂二级结构的 RNA 和不纯 RNA 的能力。
分子生物学中使用的大多数反转录酶来源于禽成髓细胞瘤病毒(AMV)或莫洛尼鼠白血病病毒(MMLV)的 pol 基因。AMV 反转录酶是实验室中首批用于 cDNA 合成的酶之一。该酶是一种分子量为 170-kDa 的异二聚体,最佳反应温度范围为42-48℃。AMV 反转录酶具有较强的 RNase H 活性,可降解 RNA:cDNA 复合物中的 RNA,获得的 cDNA 片段较短(<5 kb)。
MMLV 反转录酶因具有单体结构而成为常用的替代品,可对重组酶进行更简单的克隆和修饰。MMLV 反转录酶是一种分子量为75 kDa 的酶,反应温度约为37°C。虽然 MMLV 的热稳定性低于 AMV 反转录酶,但其 RNase H 活性较低,因此具有更高的长 cDNA(<7kb)合成效率[3]。
为了进一步改善 cDNA 合成,MMLV 反转录酶经过改造具有更低的 RNase H 活性(即 RNase H Minus,将 RNase H 结构域进行突变)、更高的热稳定性(高达 55℃)和更强的持续合成能力(65 倍以上)。这些属性改变可增加 cDNA 长度和产量,提高灵敏性,改善抑制剂耐受性,并缩短反应时间(表 2)[4]。
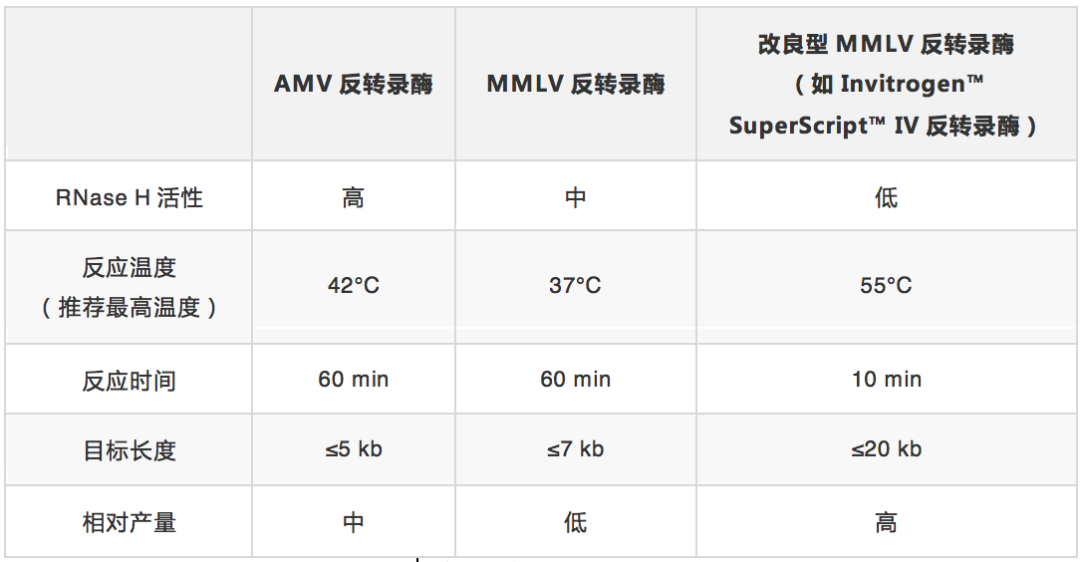 表 2. 常用反转录酶及其属性
表 2. 常用反转录酶及其属性
四、引物选择
为了启动反转录,反转录酶需要一种短 DNA 寡核苷酸(即引物)与其互补序列在 RNA 模板上结合,并作为新链合成的起点。根据 RNA 模板和下游应用,有三种基本类型的引物可供选用:oligo(dT)引物、随机引物和基因特异性引物(图 3)。
 图 3. 反转录中的常用引物。
图 3. 反转录中的常用引物。
❖ Oligo(dT)引物由 12-18 个脱氧胸腺核苷酸组成,可与真核 mRNA 的 poly(A)尾退火。其为从真核生物 mRNA 构建cDNA 文库、全长 cDNA 克隆和 cDNA 3'末端快速扩增(3'RACE)的最佳选择。由于 oligo(dT)引物对 poly(A) 尾的特异性,其不适用于降解的 RNA,如来自FFPE样品的 RNA,也不适用于缺少 poly(A)尾的 RNA,如原核生物 RNA 和 microRNA。由于 cDNA 合成开始于 3' poly(A)尾,oligo(dT)引物可能引起 3' 末端偏差。具有复杂二级结构的 RNA 也可能影响全长 cDNA 合成,导致 5' 末端的代表性偏低。
通过修饰 Oligo(dT)引物,可以提高反转录效率。例如,Oligo(dT)引物的长度可以延伸到 20 个核苷酸或更长,使其在较高温度下退火。在一些情况下,Oligo(dT)引物可能包括 3' 末端的多义碱基,如 dN(dA,dT,dG或dC)和 dV(dG,dA或dC)。该修饰可防止 poly(A)滑移,并且将起始位点锁定在 poly(A)尾的上游位置。这些引物被称为锚定oligo(dT)。
❖ 随机引物是具有随机碱基序列的寡核苷酸,通常由 6 个核苷酸组成,被称为随机六聚体、N6 或 dN6。由于随机引物的随机结合(即没有模板特异性),其可以退火到样本中的任何类型 RNA。因此,这些引物被认为可以用于无 poly(A)尾结构(如 rRNA、tRNA、非编码 RNA、小 RNA、原核 mRNA)的 RNA、降解 RNA(如来自 FFPE 组织的 RNA)和具有已知二级结构的 RNA(如病毒基因组)的反转录。
虽然随机引物有助于改善 cDNA 合成,但它们不适合用于长 RNA 的全长反转录。增加随机六聚体引物的浓度可提高 cDNA产量,但同时也会增加相同模板上与多个位点的结合,从而导致 cDNA 片段较短(图 4)。
此外,随机引物可能不适用于某些 RT-PCR 应用。例如,高估 mRNA 拷贝数就是一个需要考虑的问题[5]。两步法RT-PCR经常使用 oligo(dT)和随机引物的混合物,结合每种类型引物的优点。在 microRNA(miRNA)表达检测中,随机六聚体并不适用,必须为 miRNA 的反转录设计特殊引物[6,7]。
 图4. cDNA 的长度和产量受到所选择引物和引物浓度的影响。使用 oligo(dT)引物和不同浓度的随机六聚体引物,将具有 poly(A)尾的 6.4 kb RNA 反转录成双链 cDNA。通过琼脂糖凝胶电泳分析结果可以看到,使用 oligo(dT)引物可获得更分散、更长的转录产物,相比之下,增加随机六聚体的浓度,可产生更高浓度的短 cDNA。
图4. cDNA 的长度和产量受到所选择引物和引物浓度的影响。使用 oligo(dT)引物和不同浓度的随机六聚体引物,将具有 poly(A)尾的 6.4 kb RNA 反转录成双链 cDNA。通过琼脂糖凝胶电泳分析结果可以看到,使用 oligo(dT)引物可获得更分散、更长的转录产物,相比之下,增加随机六聚体的浓度,可产生更高浓度的短 cDNA。
❖ 基因特异性引物可提供特异性最强的反转录引物配对。这些引物是根据目标 RNA 的已知序列进行设计的。由于引物与特异性 RNA 序列结合,每个目标 RNA 都需要一套新的基因特异性引物。因此,当对多种目标进行分析时,则需要使用更多的 RNA 样本。基因特异性引物通常用于一步 RT-PCR 实验应用。
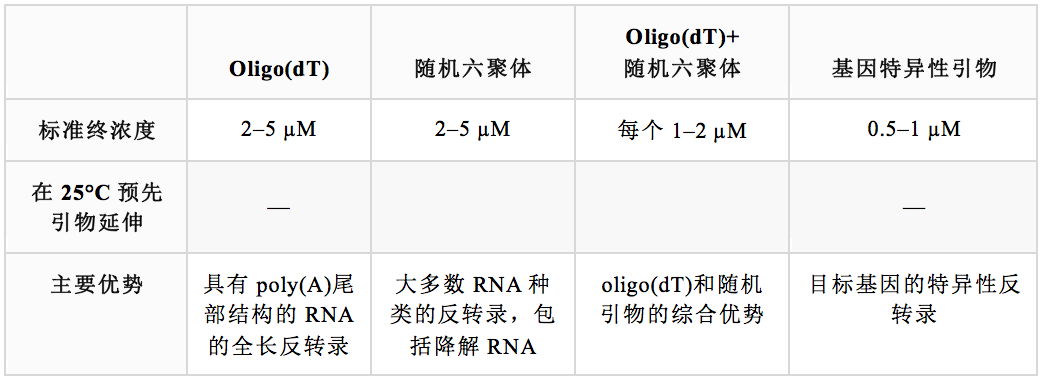 表 3. 常用反转录引物的对比
表 3. 常用反转录引物的对比
五、主要反应组分
除了反转录酶和引物之外,其它主要反应组分还包括RNA模板(预处理以去除基因组 DNA)、缓冲液、dNTP、DTT、RNA 酶抑制剂和 RNase-free 水(图 5)。
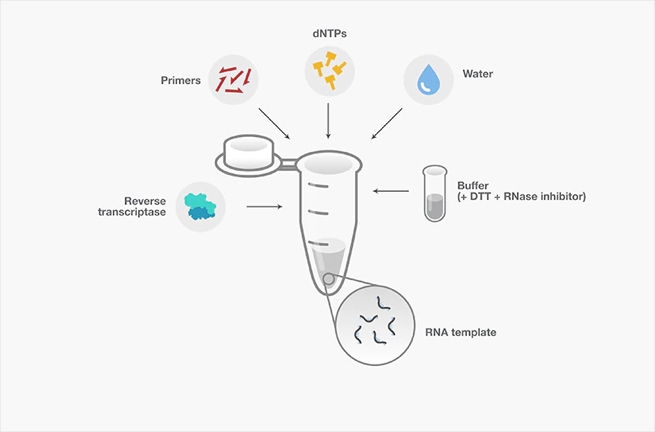 图 5. 反转录反应及其主要组分。
图 5. 反转录反应及其主要组分。
❖ RNA 模板按本文前述方法制备。表4提供了反转录反应的推荐 RNA 起始量范围,最佳起始量取决于目标序列的普遍性和反转录酶的灵敏性。
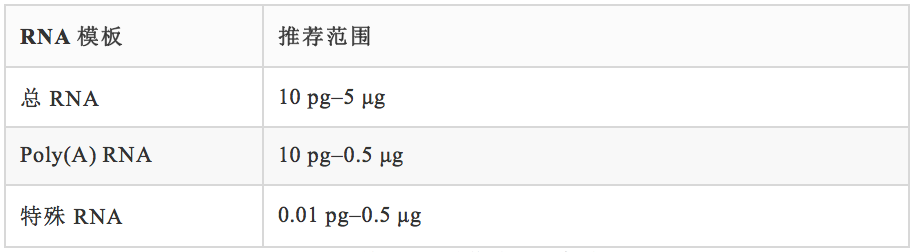 表 4. 反转录反应的推荐RNA起始量范围
表 4. 反转录反应的推荐RNA起始量范围
❖ 反应缓冲液可维持反应的最佳 pH 和离子强度。提供的缓冲液还可能含有提高反转录效率的添加剂。
❖ dNTPs 通常为 0.5-1 mM,最好为等摩尔浓度。推荐使用新鲜稀释的高品质 dNTP,以确保良好的反转录效果。
❖ DTT,一种还原剂,通常用于提供最佳的酶活性。如果 DTT 或其他添加剂发生沉淀,会降低反应效率;因此,应溶解反应组分并充分混匀。
❖ RNA 酶抑制剂通常包含在反应缓冲液中或单独被添加到反转录反应中,用于防止 RNA 降解。RNase 可能在 RNA 分离过程中被共同纯化,或者在反应体系配制期间被引入。已知的 RNase 有许多种,应根据其作用方式和反应要求选择合适的 RNA 酶抑制剂。
❖ 反转录反应中使用的水应不含有核酸酶。最好选用商业化供应的无核酸酶水,或经 DEPC(焦碳酸二乙酯)处理去除 RNase 的水。污染 RNase 不能通过简单的过滤去除,而且由于 RNase 是热稳定的,无法通过高压消毒去除。
六、反应温度和反应时间
反转录反应包含三个主要步骤:引物退火、DNA 聚合和酶失活(图 6)。这些步骤的温度和持续时间因选择的引物、目标 RNA 和使用的反转录酶而异。
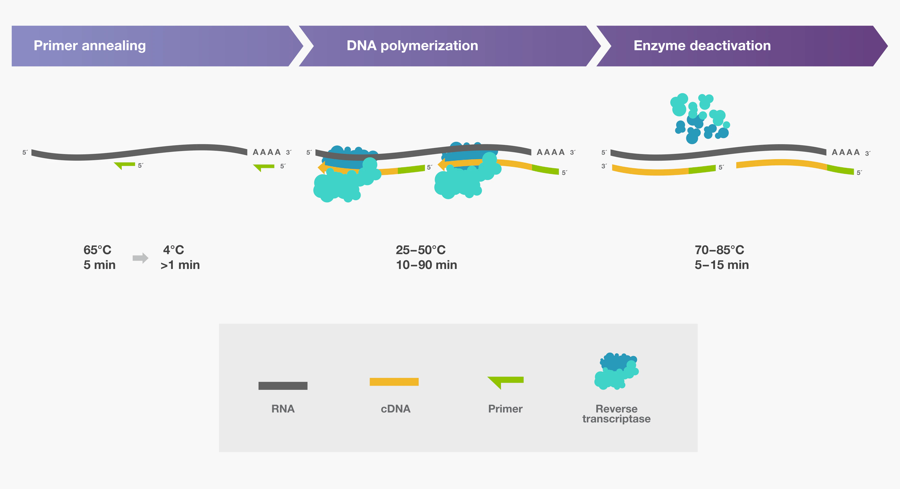 图 6. cDNA 合成的三个主要步骤。
图 6. cDNA 合成的三个主要步骤。
❖ 引物退火:将引物与 RNA 模板混合,加热至 65℃ 并维持 5 分钟,然后冰浴至少 1 分钟。这有助于确保 RNA 保持单链以及引物与靶标有效退火。退火后,加入反转录酶和必需组分(如缓冲液、dNTPs、RNA 酶抑制剂)。
❖ DNA 聚合:在此步骤中,反应温度和持续时间可能会根据所使用的引物和反转录酶而变化。使用 oligo (dT)引物(Tm〜35-50 ℃),可以在反转录酶的最佳反应温度(37-50 ℃)下直接孵育反应。随机六聚体引物因其较短的长度,通常具有较低的 Tm(〜10-15℃)。因此,当使用随机六聚体引物(单独使用或与 oligo(dT)结合使用)时,我们建议在加入酶后,在室温(〜25℃)孵育 10 分钟以延伸引物。
反转录酶的热稳定性各不相同,而热稳定性决定了每种酶的最佳聚合温度。使用热稳定的反转录酶,可达到更高的反应温度(如 50 ℃),有助于高GC或具有复杂二级结构 RNA 的变性,同时不影响酶活性(图 7)。
聚合时间取决于反转录酶的持续合成能力,即单次合成过程中掺入的核苷酸数量。例如,持续合成能力较低的野生型 MMLV 反转录酶通常需要 60 多分钟才能完成 cDNA 合成。相比之下,持续合成能力较高的改良型反转录酶可能只需要 10 分钟时间,就可以合成 9 kb cDNA。
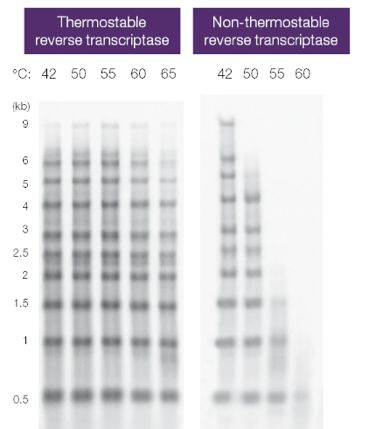 图 7. 热稳定性对反转录酶活性的影响。使用 oligo(dT)引物和放射性标记 dNTPs,对含有不同长度 RNA 的样本进行反转录。通过凝胶电泳分离反应产物,并通过放射自显影进行显色。热稳定的反转录酶即使在 50℃ 以上也能得到高 cDNA 产量。
图 7. 热稳定性对反转录酶活性的影响。使用 oligo(dT)引物和放射性标记 dNTPs,对含有不同长度 RNA 的样本进行反转录。通过凝胶电泳分离反应产物,并通过放射自显影进行显色。热稳定的反转录酶即使在 50℃ 以上也能得到高 cDNA 产量。
❖ 酶失活:反转录反应的最后一步是将反转录酶失活处理。失活温度范围为 70-85 ℃,具体取决于酶的热稳定性。失活通常需要 5-15 分钟,温度越高,所需时间越短。
七、第一链和第二链 cDNA 合成
如前所述,从 RNA 模板合成 cDNA,产生 cDNA:RNA 复合物。该过程被称为第一链 cDNA 合成。如果存在 RNase H 活性(如野生型 AMV 和 MMLV 反转录酶),则 cDNA:RNA 复合物中的 RNA 会在第一链合成期间被切割。第一链 cDNA 可以直接用于 RT-PCR 等实验应用中,热稳定 DNA 聚合酶(如 Taq DNA 聚合酶)将复制 cDNA 的互补链。
在 cDNA 文库构建和测序中,将第一链 cDNA 作为模板,获得代表 RNA 靶标的双链 cDNA。这个过程被称为第二链cDNA 合成。在第二链 cDNA 合成中,推荐使用 RNase H 活性最小的反转录酶,从而最大限度地增加 cDNA 合成长度和产量。
双链 cDNA 的合成可以使用多种 DNA 聚合酶(如 T7 DNA 聚合酶、DNA 聚合酶 I、Taq DNA 聚合酶)生成 cDNA 第一链的互补链。以下列举的一些其它酶也可按照 Gubler-Hoffman 改良方法[8](图 8)用于双链 cDNA 合成。
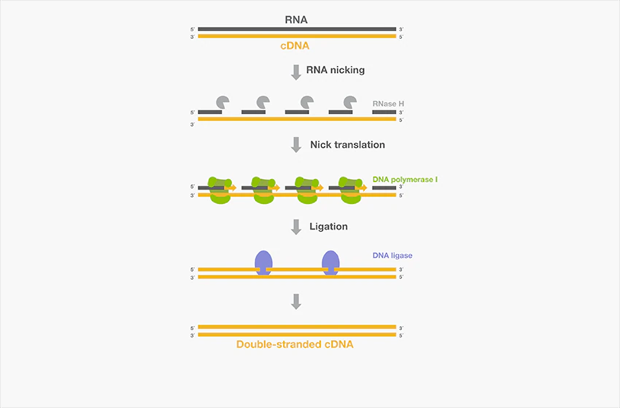 图 8. 利用 Gubler-Hoffman 方法进行双链 cDNA 合成。
图 8. 利用 Gubler-Hoffman 方法进行双链 cDNA 合成。
❖ E. coli RNase H 切割 cDNA:RNA 复合物中的 RNA 链,为 DNA 合成提供 3'-OH 引物结合位点
❖ E. coli DNA polymerase I 利用 5′-3′' 聚合酶活性延伸切口 RNA 链,并利用 5'-3' 外切核酸酶活性沿合成方向替换 RNA 链,该过程被称为切口平移。
❖ E. coli DNA ligase 缝合新合成 cDNA 片段之间的切口。(T4 DNA 连接酶不能用作替代物,因为它会连接钝端双链 cDNA 片段并形成嵌合结构。)
❖ T4 DNA polymerase 可将双链 cDNA 的末端平端化(最后一步,可选)。
总之,反转录实验的成功高度依赖于反应组分和反应条件。以上为关于反转录的一些总结,希望能给大家的实验带来些许帮助。最后,祝大家实验顺利!
参考文献:
1.Schroeder A, Mueller O, Stocker S et al. (2006) The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7:3.
2.Mueller O, Lightfoot S, Schroeder A (2016) RNA Integrity Number (RIN) – Standardization of RNA Quality Control. Agilent Technologies Publication 5989-1165EN.
3.Invitrogen Corp. (2002) High performance RT for reliability in every experiment. (Brochure)
4.Thermo Fisher Scientific (2015) SuperScript IV Reverse Transcriptase. (White paper)
5.Zhang J, Byrne CD (1999) Differential priming of RNA templates during cDNA synthesis markedly affects both accuracy and reproducibility of quantitative competitive reverse-transcriptase PCR. Biochem J 337 (Pt 2):231–241.
6.Chen C, Ridzon DA, Broomer AJ et al. (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33(20):e179.
7.Kramer MF (2011) Stem-loop RT-qPCR for miRNAs. Curr Protoc Mol Biol Unit 15.10.
8.Gubler U, Hoffman BJ (1983) A simple and very efficient method for generating cDNA libraries. Gene 25(2-3):263-269.