DNA测序技术
互联网
在分子生物学研究中,DNA 的序列分析是进一步研究和改造目的基因的基础。目前用于测序的技术主要有Sanger等(1977)发明的双脱氧链末端终止法和Maxam和 Gilbert(1977)发明的化学降解法。这二种方法在原理上差异很大,但都是根据核苷酸在某一固定的点开始,随机在某一个特定的碱基处终止,产生A,T,C,G四组不同长度的一系列核苷酸,然后在尿素变性的PAGE胶上电泳进行检测,从而获得DNA 序列。目前Sanger测序法得到了广泛的应用。
Sanger 法测序的原理就是利用一种DNA 聚合酶来延伸结合在待定序列模板上的引物。直到掺入一种链终止核苷酸为止。每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。由于ddNTP缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。终止点由反应中相应的双脱氧而定。每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。
第一节 非同位素银染测序系统操作技术
一、概述:
Promega公司的SILVER SEQUENCETM DNA 测序系统是一种无放射性的序列分析系统,它通过灵敏的银染方法检测凝胶中的条带。 银染提供了一种对于放射性或荧光法来说更加快速,廉价的替代方法。测序结果可以在同一天内得到;电泳完成后经90分钟就可读序,这是常规的放射性测序法做不到的。 此外,SILVER SEQUENCETM系统用未修饰的5'OH寡聚核苷酸作为引物, 减少了特殊修饰寡聚核苷酸的花费。该系统不需要放射性方法中对同位素的谨慎操作,也不需要荧光法或化学发光技术的昂贵试剂。另外,也不需要象大多数荧光法那样用仪器来检测序列条带。
Taq DNA 聚合酶在95℃时极强的热稳定性。本系统利用的测序级Taq DNA 聚合酶是一种Taq DNA 聚合酶的修饰产品,对于双链DNA 模板有非常好的效果,具有高度的准确性,能产生均一的条带,且背景低。
SILVER SEQUENCETM系统包含被修饰的核苷酸混合物,如7-去氮dGTP(7-deaza dGTP,或dITP)替代dGTP可清除由GC丰富区域所引起的条带压缩现象。
退火温度是热循环测序中最重要的因素。高退火温度可减少模板二级结构。提高引物结合模板配对的严谨性。链重退火和模板二级结构则限制了小片断PCR产物(<500bp)得到清楚的序列数据的能力。引物延伸起始于每个循环的退火阶段。在较低温度时,聚合酶可能会遇到坚固的二级结构区域,它可导致聚合酶解离。则在四个电泳道中均有同一相对位置的条带。因为这些原因,应该使用尽可能高的退火温度。对于有牢固二级结构的模板建议使用95℃变性、70℃退火/延伸的循环模式。一般来说,较长的引物及GC含量高的引物能得到较强的信号。实验结果表明,>24mer的GC含量约为50%的引物可得到最佳结果。
由于本系统采用热循环装置,与常规的测序方法相比具有如下几点好处:(1).本方法线性扩增模板DNA 产生足够的产物使银染技术能够检测序列条带,测序反应需要0.03--2pmol模板DNA ,随模板种类而定。(2). 在每一个变性循环中的高温可以取代对双链DNA (dsDNA )模板的碱变性及乙醇沉淀过程,变性循环也有助于消除由于线性dsDNA 模板(如PCR反应产物)快速重退火所引起的问题。(3). 高温聚合酶反应减弱了DNA 模板的二级结构,允许聚合酶穿过高度二级结构化的区域。
二、材料 待测已提纯的DNA ,可为单链,也可为双链。
三、设备 高压电泳仪,测序用电泳槽,制胶设备,PCR仪。
四、试剂
(1)SILVER SEQUENCETM DNA 测序试剂盒。
(2)丙烯酰胺和甲叉双丙烯酰胺储备液(38%丙烯酰胺 W/V,2%甲叉双丙烯酰胺 W/V):95g丙烯酰胺,5g甲叉双丙烯酰胺溶于140ml 双蒸水中,定容至250ml,0.45mm过滤器过滤后,贮于棕色瓶中,置于4℃冰箱可保存2周。
(3)10%过硫酸铵,0.5g过硫酸铵溶于4ml水中,定容至5ml,应新配新用。
(4)10×TBE缓冲液(1 mol/L Tris,0.83mol/L 硼酸,10mmol/L EDTA): 121.1g Tris,51.35g硼酸,3.72g Na2 EDTA ・2H2O,溶于双蒸水中定容至1升,置于4℃下可贮存2周,其pH约为8.3。
(5)TBE电极缓冲液:10×TBE 缓冲液稀释至1×TBE备用。
(6)TEMED
(7)固定/停止溶液:10%冰醋酸(V/V)配制2升备用。
(8)染色溶液:硝酸银2克,甲醛3ml,溶于2升超纯水中备用。
(9)显影溶液:60克碳酸钠(Na2CO3)溶于2升超纯水中,使用前加3ml 37% 甲醛和 40ml硫代硫酸钠溶液(10mg/ml)。
(10)95%乙醇。
(11)0.5%冰乙酸。
(12)Sigmacote (Sigma CAT. #SL-2)。
五、操作步骤:
成功地使用银染测序系统需要对提供的操作方法进行仔细考虑。银染不如放射性检测法灵敏,而需要更多的模板量,此外,也不可能通过延长X-光胶片曝光时间的方法增加信号强度。因此,请使用推荐的DNA 模板量,每次均使用所提供的对照检查系统的可靠性,并且注意如下几点:
(1) DNA 的浓度和纯度必须经过琼脂糖凝胶电泳或荧光法测定,样品应与已知量DNA 一起电泳。
(2) 分光光度法对于很多DNA 提取物包括质粒小量制备来说,并不能给出一个可信的DNA 浓度估计,混杂的染色体DNA 、蛋白、RNA、有机物及无机化合物均可能有260 nm光吸收。因此,分光光度法常常错误地高估DNA 浓度。
(3) DNA 制备过程中用核糖核酸酶处理所产生核糖核苷酸,虽然它们在电泳后DNA 样品的前面,并不能观察到,但它们仍会有260nm光吸收。
(一)测序反应:
1. 对于每组测序反应,标记四个0.5ml eppendorf管(G、A、T、C)。每管加入2ml适当的d/ddNTP混合物(d/ddNTP Mix)。各加入1滴(约20ml)矿物油,盖上盖子保存于冰上或4℃备用。
2. 对于每组四个测序反应,在一个eppendorf管中混合以下试剂:
(1) 样品反应:
质粒模板DNA 2.1pmol
5×测序缓冲液 5ml
引物 4.5pmol
无菌ddH2O 至终体积16ml
(2)对照反应
pGEM-3Zf(+)对照DNA (4mg) 4.0ml
5×测序缓冲液 5ml
pUC/M13正向引物(4.5pmol) 3.6ml
无菌ddH2O 至终体积 16 ml
3. 在引物/模板混合物(以上第2步)中加入1.0ml测序级Taq DNA 聚合酶(5u/ml)。用吸液器吸动几次混匀。
4. 从第3步的酶/引物/模板混合物中吸取4ml加入每一个d/ddNTP混合物的管内。
5. 在微量离心机中离心一下,使所有的溶液位于eppendorf管底部。
6. 把反应管放入预热至95℃的热循环仪,以[注意]中循环模式为基准,开始循环程序。对于每个引物/模板组合都必须选择最佳退火温度。下列程序一般能读出从引物开始350碱基的长度。
7. 热循环程序完成后,在每个小管内加入3μl DNA 测序终止溶液,在微量离心机中略一旋转,终止反应。
[注意] 1、测序所用模板DNA 的量一般按下面要求加入:
| 模板种类/长度 | 模板量 |
| 200bp (PCR产物) | 16ng(120fmol) |
| 3000-5000bp(超螺旋质粒DNA ) | 4mg (2pmol) |
| 48000bp(λ,粘粒DNA ) | 1mg(31fmol) |
由于超螺旋质粒产生的信号比松驰的线性双链DNA 弱,因此使用超螺旋质粒作为模板时其用量要比其它模板大一些。
2、计算与4.5pmol相当的引物纳克数可用以下一般公式:
4.5pmol=1.5ng×n,其中n为引物碱基数
计算与1pmol相当的引物微克数可用以下一般公式:
dsDNA :1pmol=(6.6×10-4 mg)×n,其中n为模板碱基对数
ssDNA :1pmol=(3.3×10-4 mg)×n,其中n为模板碱基数
3、为阻止Taq DNA 聚合酶延伸非特异性退火引物, 热循环仪必须预热至95℃。温度变换应越快越好。下面的循环时间不包括变温时间。如果你无法确定使用何种模式,建议从模式1开始。
模式1:适用于引物<24碱基或GC含量<50%
95℃ 2分钟。然后: 95℃ 30秒(变性),42℃ 30秒(退火),70℃ 1分钟(延伸)。
模式2:适用于≥24碱基或略短的GC含量≥50%的引物。
95℃ 2分钟,然后: 95℃ 30秒(变性),70℃ 30秒(退火/延伸)。 4. 在加入终止溶液之后样品可在4℃保存过夜。
(二)、 测序凝胶板的制备
1、玻璃板的处理:
银染测序的玻璃板一定要非常清洁,一般先用温水和去污剂洗涤,再用去离子水冲洗玻璃板,除去残留的去污剂,最后用乙醇清洗玻璃板。玻璃板上遗留的去污剂微膜可能导致凝胶染色时背景偏高(棕色)。短玻璃板经粘合溶液处理可将凝胶化学交联于玻璃板上。这一步对于在银染操作过程中防止凝胶撕裂至关重要。
(1)短玻璃板的处理
A. 在1ml 95%乙醇,0.5%冰乙酸中加入5ml粘合硅烷(Bind Silane),配成新鲜的粘合溶液。
B. 用经浸透新配的粘合溶液浸透的吸水棉纸擦拭仔细清洗过并已经自然干燥的玻璃板,整个板面都必须擦拭。
C. 4-5分钟后,用95%乙醇单向擦玻璃板,然后略用力沿垂直方向擦拭。重复三次这一清洗过程,每次均须换用干净的纸,除去多余的粘合溶液。
[注意] 1. 在95%乙醇单向擦玻璃板时过度用力会带走过多的粘合硅烷,使凝胶不能很好地粘附。
2. 准备长玻璃板之前要更换手套,防止粘染粘合硅烷。
3、防止粘合溶液沾染在长玻璃板上是很重要的,否则将导致凝胶撕裂。
(2)、长玻璃板的处理
A. 用浸透Sigmacote溶液的棉纸擦拭清洗过的长玻璃板。
B. 5-10分钟后用吸水棉纸擦拭玻璃板以除去多余的Sigmacote溶液。
[注意] 1. 用过的凝胶可在水中浸泡后用剃须刀片或塑料刮刀刮去。玻璃板须用去污剂完全清洗。或者凝胶用10% NaOH浸泡后除去。为防止交叉污染,用于清洗短玻璃板的工具必须与清洗长玻璃板的工具分开,如果出现交叉污染,以后制备的凝胶可能撕裂或变得松驰。
2、凝胶的制备:
(1)玻璃板经粘合硅胶和Sigmacote处理后,即可固定玻璃板。该方法是用0.2mm或0.4mm厚的边条置于玻璃板左右两侧,将另一块玻璃板压于其上。在长玻璃板的一侧插入鲨鱼齿梳平的一面边缘,用夹子固定住。
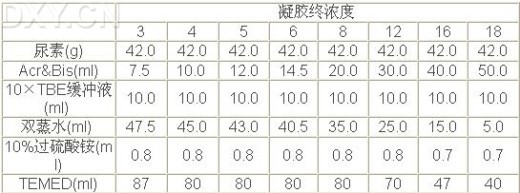
(2)根据所需要的凝胶浓度,按下表制备测序凝胶,一般6%-8%的胶浓度可获得较好的结果。配制过程中,先用适量双蒸水溶解尿素,再加入Acr&Bis和10×TBE缓冲液,再用双蒸水调终体积至99.2ml,并用0.45mm的滤膜过滤,然后加过硫酸铵和TEMED。溶解尿素时不必加热。如果确需加热则应等溶液完全冷却后,方可加入TEMED和过硫酸铵。一般在胶灌制后4-6分钟,即开始聚合,如果聚合不好,则应使用高浓度的TEMED和过硫酸铵。
| 凝胶终浓度 | ||||||||
| 3 | 4 | 5 | 6 | 8 | 12 | 16 | 18 | |
| 尿素(g) | 42.0 | 42.0 | 42.0 | 42.0 | 42.0 | 42.0 | 42.0 | 42.0 |
| Acr&Bis(ml) | 7.5 | 10.0 | 12.0 | 14.5 | 20.0 | 30.0 | 40.0 | 50.0 |
| 10×TBE缓冲液(ml) | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 |
| 双蒸水(ml) | 47.5 | 45.0 | 43.0 | 40.5 | 35.0 | 25.0 | 15.0 | 5.0 |
| 10%过硫酸铵(ml) | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 | 0.7 | 0.7 |
| TEMED(ml) | 87 | 80 | 80 | 80 | 80 | 70 | 47 | 40 |
(3)胶配制好后,即可灌制胶板。一般是将凝胶沿着压条边缘缓慢地倒入玻璃板的槽中,倒完后,静止放置使之聚合完全。
[注意] 1、使用夹子固定玻璃板时,最好夹子的力量稍大一些,防止因力量不足使灌胶的过程中出现漏胶液现象。
2、灌制凝胶的过程中要严防产生气泡,否则影响测序的结果。
(三)电泳:
1、预电泳
(1)当凝胶聚合完全后,拨出鲨鱼齿梳,将该梳子反过来,把有齿的一头插入凝胶中,形成加样孔。
(2)立即将胶板固定在测序凝胶槽中,一般测序凝胶槽的上下槽是分开的,因而只有在固定好凝胶板后,方能加入TBE缓冲液。
(3)稀释10×TBE缓冲液至1×TBE,将该缓冲液加入上下二个电泳槽中,去除产生的气泡,接上电源准备预电泳。
(4)有些电泳槽,如LKB的Macrophor等是使用水浴加热的,则应先将水浴加热至55℃后进行预电泳。有的不使用水浴加热,依靠电泳过程中自身产生的热进行保温,如上海求精有机玻璃仪器生产的测序电泳槽,这种槽需夹上二块散热铝板,使整个凝胶板的温度一致。
(5)按30V/cm的电压预电泳20-30分钟。预电泳的过程是去除凝胶的杂质离子,同时使凝胶板达到所需的温度。高温电泳可防止GC丰富区形成的发夹状结构,影响测序的结果。
[注意]
(1) 用鲨鱼齿梳制作加样孔时,应注意将齿尖插入胶中0.5mm左右,千万注意不能使加样孔渗漏,否则得不到正确的结果。
(2).应时刻注意上面电泳槽中的缓冲液是否渗漏,否则极易造成短路而损坏电泳仪。
2、样品的制备:
当在预电泳时,即可进行样品的制备,将反应完毕的样品在沸水浴中加热1-3分钟,立即置于冰上即可。如果样品长时间不用,则应重新处理。可使用4-6%聚丙烯酰胺凝胶,胶厚0.4mm。厚度小于0.4mm的胶可能导致信号太弱。加样时不必吸去上层覆盖的矿物油,但要小心地吸取矿物油下的蓝色样品。
3、上样及电泳
关闭电泳仪,用移液枪吸缓冲液清洗样品孔,去除在预电泳时扩散出来的尿素,然后立即用毛细管进样器吸取样品,加入样品孔中。上样顺序一般为G、A、T、C。加样完毕后,立即电泳。开始可用30V/cm进行电泳,5分钟后可提高至40-60V/cm,并保持恒压状态。一般来说,一个55cm长,0.2mm厚的凝胶板,在2500V恒压状态下电泳2小时即可走到底部,同时在电泳过程中,电流可稳定地从28mA降至25mA。为了能读到更长的序列,可采用两轮或多轮上样。
[注意]
1、上样电泳时,一定要注意凝胶板的温度是否达到55℃左右,如果还没有达到,则应等温度达到后才能上样电泳。
2、一般来说电泳时,不宜使用太高的电压,因为太高的电压会使凝胶的分辨率降低,并且使带扩散。电泳中可进行恒功率电泳。
(四)、 测序凝胶的银染
染色过程要求凝胶浸在塑料盘中。因而至少使用两个盘子,大小与玻璃板类似。在盘中加入新鲜溶液之前须用高质量的水洗涤盘子。
1. 电泳完毕后用一个塑料片子小心地分开两板,凝胶应该牢固地附着在短玻璃板上。
2. 固定凝胶:将凝胶(连玻璃板)放入塑料盘,用固定/停止溶液浸没,充分振荡20分钟或直至样品中染料完全消失,胶可在固定/停止溶液中保存过夜(不振荡)。保留固定/停止溶液,用于终止显影反应。
3. 洗胶:用超纯水振荡洗胶3次,每次2分钟。从水中取出,当转移至下一溶液时拿着胶板边沿静止10-20秒,使水流尽。
4. 凝胶染色:把凝胶移至染色溶液充分摇动30分钟。
5. 凝胶显影:
(1). 在显影溶液中加入甲醛(3ml)和硫代硫酸钠溶液(400μl)以完成显影液的配制。
(2). 从染色溶液中取出凝胶放入装有超纯水的盘中浸洗5-10秒。注意,把凝胶从超纯水转移到显影溶液的总时间不能长于5-10秒。浸泡时间过长则导致信号微弱或丧失信号。若浸泡时间过长,可重复第五步用染色液浸泡。
(3). 立刻将凝胶转移至1升(总量的一半)预冷的显影液充分振荡直至模板带开始显现或开始出现第一批条带,把凝胶移入剩下的1升显影液中继续显影2--3分钟,或直至所有条带出现。
6. 固定凝胶:在显影液中直接加入等体积的固定/停止溶液。停止显影反应,固定凝胶。
7. 在超纯水中浸洗凝胶两次,每次2分钟,注意在本操作中戴手套拿着胶板边缘避免在胶上印上指纹。
8. 将凝胶置于室温干燥或用抽气加热法干燥。在可见光灯箱或亮白,黄色背景(如纸)上观察凝胶,若需永久保存的记录,则可用EDF胶片保留实验结果。
[注意] 测序产物的银染是显现序列信息的一种新方法,本系统的成败受几个因素的影响。
1. 水的质量对于染色的成功极其重要。超纯水(NANOpureR 或Milli-QR 的水)或双蒸水可获得较好的效果,如果水中有杂质,则低分子量条带可能无法出现。
2. 碳酸钠也非常重要。使用新鲜的,美国化学学会级碳酸钠较好,如Fisher和Kodak ACS试剂级碳酸钠(Fisher Cat #S263-500或S262-3,或Kodak Cat #109-1990),一般可获得较好的结果。
3. 染色后的洗涤步骤是非常关键的。如果凝胶洗涤时间太长,银颗粒会脱离DNA ,产生很少或没有序列信号。如果洗涤时间过长,染色步骤可以重新进行。
4. 如果凝胶厚度超过0.4mm或丙烯酰胺浓度高于4-6%,则有必要延长固定和染色的时间。如果凝胶比0.4mm薄,染色反应后的洗涤必须缩短至不超过5秒。
5. 在室温下进行所有步骤,显影反应除外。显影溶液必须预冷至10-12℃以减小背景杂色。注意:临用前在显影溶液中加入甲醛和硫代硫酸钠。用新配的染色及显影溶液。不要重复使用任何溶液。
(五)、EDF胶片显影
使用EDF胶片可增强测序条带的对比度,如果测序胶上条带很淡,我们建议把数据转移至EDF胶片,银染胶在其影像转移至EDF胶片之后可增强条带可读性。
1. 在暗室内,将染色过的粘于玻璃板的凝胶(胶面向上)置于荧光灯箱上。如有合适的漫射板,亦可用白灯箱,为确保曝光时间,用一小条EDF胶片曝光不同时间,检查不同的曝光强度,一般曝光20--40秒可得较好结果。
2. 在红灯下找到EDF胶片有缺刻的一角,然后把胶片置于凝胶上,使缺口位于左上角。由于EDF胶片是单面的,因此必须确保缺口在左上角。
3. 在EDF胶片上放置一干净、干燥的玻璃板,开亮灯箱约20秒。
4. 用冲显放射自显影胶片的步骤手工显影EDF胶片,可使用下列操作过程:
a. 在Kodak GBX显影液中显影1-5分钟;
b. 水洗1分钟;
c. 在Kodak GBX定影液中定影3分钟;
d. 水洗1分钟.
[注意] 1. 进行EDF胶片显影之前凝胶必须完全干燥。戴手套进行操作,以免印上指纹。同时注意EDF胶片不能用自动胶片处理器。
2. 对于不同的光源最佳曝光时间可能不同,通过对一小条EDF胶片曝光不同时间以选择你所用光源的最佳曝光时间,参阅胶片说明书。
3. 曝光时间短则EDF胶片影像较深,曝光时间长则有助于减弱背景。
第二节 T7 DN A聚合酶测序技术
一、概述
T7 DNA 聚合酶最初具有5'→3'聚合酶活性以及单链和双链3'→5'外切酶活性。当T7 DNA 聚合酶用适当方法处理后,可使3'→5'外切酶活力明显下降。改造后的T7 DNA 聚合酶又称T7测序酶。
使用T7测序酶得到的测序数据具有在每个碱基位置都有相对均匀的配对参入。因此,放射自显影所得到的结果较为清晰易辩,对于许多模板来说,使用含有dGTP的混合物即可得到理想的测序结果。然而,如果出现了带压缩的问题,则用含dITP的混合物来取代常规的dGTP。dITP的掺入,使在富含GC的模板中也能有效地消除带压缩现象。
T7测序酶具有很高的聚合速度,并有高度的延续性,正因如此,该酶在使用中不同于Klenow片段和反转录酶。它几乎没有错误性的终止,整个反应在很短的时间内完成,并且不需要进行追加反应(Chase step)。 然而对于有紧密二级结构的区域,错误的终止反应会给阅读序列带来困难。如果碰到这些情况,应使用一些高温DNA 聚合酶,如Promega公司的测序级Taq DNA 酶。利用高温DNA 聚合酶独有的耐热特性,测序反应可在不利于形成二级结构的高温条件下进行。
T7 DNA 聚合酶测序的快速而简便的方法是从Klenow酶和AMV反转录酶测序的操作步骤修改而来的,它的最大优点是具有很高的5'-3'的DNA 合成活性和极低的3'-5'端外切酶的活性。在测序过程中,若以同位素标记则相对于大肠杆菌DNA 聚合酶Ⅰ的大片段Klenow,或反转录酶AMV而言,则可使条带非常的均一,并且它的放射性背景极低。
T7 DNA 聚合酶测序时DNA 的合成的过程是分二步完成的。第一步为标记反应阶段,第二步方为双脱氧链末端终止的DNA 合成过程。在第一步过程中,引物的延伸是在较低浓度的脱氧三磷酸核苷酸的存在下完成的,在此反应过程中,已含有经放射性标记的dATP。第二步过程中,脱氧三磷酸核苷酸浓度提高,并且加入双脱氧的三磷酸核苷酸,DNA 在合成过程中,因加入的双脱氧三磷酸核苷酸而随机终止,形成长短不一的DNA 片段。
二、材料待测的DNA 模板,可用双链或单链模板。
三、设备高压电泳仪,测序用电泳槽,照相显影用大号塑料盆,制胶设备,吹风机,放射自显影盒,X-光片。
四、试剂
1、5×T7 DNA 聚合酶缓冲液:200mmol/L Tris・Cl,pH7.5,100mmol/L MgCl2,250mmol/L NaCl。
2、引物:使用通用引物pUC/M13正向或逆向引物。
3、DTT : 0.1mol/L。
4、5×常规标记混合物:7.5mmol/L dGTP,7.5mmol/L dCTP,7.5mmol/L dTTP。
5、5×dITP标记混合物:15mmol/L dITP,7.5mmol/L dCTP,7.5mmol/L dTTP。
6、dNTP A溶液(适用于dGTP): 80mmol/L dGTP,80mmol/L dATP,80mmol/L dCTP,80mmol/L dTTP,50mmol/L NaCL。
7、ddG终止混合物 (适用于dGTP):dNTP A溶液再加 8mmol/L ddGTP。
8、ddA终止混合物 (适用于dGTP):dNTP A溶液再加 8mmol/L ddATP。
9、ddT终止混合物(适用于dGTP):dNTP A溶液再加 8mmol/L ddTTP。
10、ddC 终止混合物(适用于dGTP):dNTP A溶液再加 8mmol/L ddCTP。
11、dNTP B溶液(适用于dITP): 160mmol/L dITP,80mmol/L dATP,80mmol/L dCTP,80mmol/L dTTP,50mmol/L NaCl。
12、ddG终止混合物(适用于dITP):dNTP B溶液再加1.6mmol/L ddGTP。
13、ddA 终止混合物(适用于dITP):dNTP B溶液再加1.6mmol/L ddATP。
14、ddT 终止混合物(适用于dITP):dNTP B溶液再加1.6mmol/L ddTTP。
15、dd C终止混合物(适用于dITP):dNTP B溶液再加1.6mmol/L ddCTP。
16、测序用T7 DNA 测序酶。
17、酶稀释缓冲液:10mmol/L Tris・HCl,pH7.5,5mmol/L DTT,0.5mg/ml BSA 。
18、终止溶液: 95%甲酰胺,20mmol/L EDTA,0.05%溴酚兰,0.05%二甲苯兰 。
19、[a-32P]dATP 或[a-35S]-dATP,35S的优点是可使条带为窄而清晰,并且操作更为安全。放射强度为:1000-1500Ci/mmol,10mCi/ml。
20、TE缓冲液:10mmol/L Tris-HCl,1mmol/L EDTA,pH7.5。
21、上节所述的凝胶制备过程中的全套试剂。
22、X光显影液:H2O:50℃ 800ml,米吐尔 2.2g,无水Na2SO3 72g,对苯二酚 8.8g,无水NaCO3 48g ,KBr 4g, 定容至1000ml备用。
23、F-5坚膜定影液:水600ml ,Na2S2O3 240g,Na2SO3 15g,冰醋酸 13.4ml;硼酸 7.5g ,粉状钾矾 15g ,定容至1000ml 备用。
24、TYP肉汁培养基:16g蛋白胨,16g酵母提取物,5g NaCl,2.5g K2HPO4,加水溶解,定容至1000ml。
25、20%PEG/3.75mol NH4Ac 溶液:40% PEG(分子量8000)储备液和7.5mol/L pH7.2 NH4Ac储备液等体积混合。
五、操作步骤:
(一)、模板制备
1、M13单链模板制备:在转化入合适的大肠杆菌宿主菌且在含有指示剂如X-gal/IPTG的培养基上铺板之后,含有M13重组子的细胞将表现出无色的"噬菌斑",事实上受感染的细胞并非被噬菌体溶菌或杀死,出现噬菌斑是因为被感染的细菌在生长速度上比其周围未感染的细菌慢的缘故,从无色噬菌斑上得到的感染细胞经培养便能产生测序反应所需的单链模板。
(1)过夜培养的宿主细胞(如NM522或JM101)用3ml TYP肉汁培养基以1:100稀释。在37℃强烈震荡1小时之后,细胞进入对数早期。此时,将一个合适的含有重组M13的噬菌斑(连同琼脂)用滴管转入该细胞培养液中。
(2)37℃强烈震荡(300rpm) 培养6小时。
(3)把细胞培养液转入两只1.5ml eppendorf管,12000g离心15分钟。上清液转移到新的eppendorf管再离心15分钟。小心地取出1-1.2ml上清液(注意不能触及沉淀),再转到新的eppendorf管。 .
(4)将0.25体积的含20%PEG(-8000)的3.75M醋酸铵(pH7.5)加入上清液以沉淀噬菌体。混匀后置冰上30分钟,再以12000g离心15分钟,倾去上清液,再以同样方法离心一次,用吸液器尖头将残留的PEG充分吸干净。此时应能见管底有芝麻大小的噬菌体颗粒沉淀。
(5)将沉淀物重溶于400ml TE缓冲液中。
(6)加等体积氯仿/异戊醇(24:1)混合液,强烈震荡1分钟,12000g离心5分钟。
(7)将水相转入一新的eppendorf管,注意不能触动原管中两相交界面。加等体积苯酚/氯仿(1:1)混合液,强烈振荡1分钟,12000g离心5分钟。
(8)将上层水相转入另一新的eppendorf管,重复第7步的提取,如有必要重复多次直至二相之间无可见物质存在。
(9)将上层水相转入一新的eppendorf管,加等体积氯仿,强烈振荡1分钟后,12000g离心5分钟,多次重复此步骤。
(10)将上层水相转入新的eppendorf管,加0.5倍体积的7.5 mol/L 醋酸铵,2倍体积乙醇,混匀后-20℃放置 30分钟。
(11)12000g离心15分钟,去除上清液,用70%乙醇小心清洗沉淀,如果沉淀已被搅起,重新离心,倾去酒精,真空干燥沉淀物。
(12)将DNA 的沉淀物溶解于20ml去离子水中,取2ml样品进行琼脂糖凝胶电泳定量,如果DNA 量充足,则可进行退火测序。
2、噬粒(Phagemid)单链模板的制备:
噬粒是指含有丝状单链DNA 噬菌体复制区的嵌合质粒。分子克隆中常用的pBluescript系列和pGEM系列的质粒均属噬粒。含重组噬粒的细胞单菌落经液体培养并用辅助噬菌体超感染后就能产生测序反应所需的单链DNA 模板。
(1)从新鲜平板上挑得含pGEM质粒DNA 的抗Amp单菌落,并接种到100ml含有50mg/ml氨苄的TYP肉汤培养基中,在37℃振荡过夜。
(2)取100ml过夜培养物接种到另一新的装有5ml 含50mg/mlAmp的TYP肉汁培养基的试管中,在37℃强烈振荡30分钟。
(3)加40μl辅助噬菌体R408或M13 K07,继续剧烈搅拌振荡培养6-8小时,使辅助噬菌体超感染。
(4)12000g离心15分钟后,去除沉淀,取上清,转移到一新eppendorf管中再次离心15分钟,小心地取1-1.2ml上清液(注意不能触及沉淀)。
(5)将0.25体积的含20% PEG-的3.75M醋酸铵加入上清液以沉淀噬菌体颗粒。混匀后置冰上30分钟,再以12000g离心15分钟,倾去上清液,再以同样方法离心一次,用移液器尖头将残留的PEG溶液吸净。此时应能看到管底有芝麻大小的噬菌体颗粒沉淀。
(6)将沉淀物溶解于400ml TE缓冲液(10mmol/L Tris-HCl pH8.0,1mmol/L EDTA)。
(7)加等体积氯仿/异戊醇(24:1)混合液,强烈振荡1分钟,再以12000g离心5分钟。
(8)将水相转入一干净eppendorf管(不要触动原管中两相交界面),加等体积酚/氯仿(1:1)混合液,强烈振荡1分钟,12000g离心5分钟。
(9)上层水相转入一干净eppendorf管重复第8步骤提取,如有必要重复多次直至二相之间无可见物质。
(10)将上层水相转入一干净eppendorf管加等体积氯仿,强烈振荡1分钟后离心,多次重复此步骤。
(11)将上层水相转入一干净eppendorf管加0.5倍体积7.5mol/L pH7.5醋酸铵,再加2倍体积乙醇,混和后-20℃放置30分钟。
(12)12000g离心15分钟,去除上清液,用70%乙醇小心清洗沉淀,如沉淀已被搅起,重新离心,倾去乙醇,真空干燥沉淀。
(13)将沉淀物溶解于20ml去离子水中,取2ml样品进行琼脂糖凝胶电泳进行定量,如果DNA 量充足,则进行退火和测序。
3、质粒膜板制备:参照第一章所述的方法。
(二)超螺旋质粒DNA 的碱变性:
1、取4mg(约2pmol)超螺旋质粒DNA 至一eppendorf管中,加无离子水到终体积18ml。
2、加2ml 2mmol/L NaOH/2mmol/L EDTA 溶液,置于室温(25℃下)保温5分钟。
3、加2ml 2mol/L NaAc溶液(pH4.6)涡旋混匀,使之中和。
4、加75ml无水乙醇,充分混匀后,置于干冰或-70℃沉淀10分钟。
5、于12000g离心10分钟,弃去上清,用200ml预冷的70%乙醇洗沉淀后,干燥沉淀,溶于20ml无离子水中。
(三)、测序反应:
1、引物和模板的退火:
(1)在一离心管中,混合如下几种试剂:
引物 约1-2pmol
5×T7 DNA 聚合酶测序反应缓冲液 2ml
DNA 约1mg
加ddH2O终体积至10ml 。
(2)将试管盖盖上后,65℃ 加热2分钟,然后在大约30分钟左右的时间内,使试管的温度缓慢地降到室温。降温的过程中可使用小的加热板或小型的保温仪,也可使用已调至65℃的水浴,使它们的温度在室温下缓慢地降至室温即可。当温度降至30℃以下时,退火结束。可将小试管置于冰上,退火完毕的模板须在4小时内使用。
2、标记反应
(1)在一个标准的反应过程中(从引物处开始,可读到500个碱基以上),首先需要按1:5稀释标记混合物。如4ml 混合物则加双蒸无离子水16ml。该稀释液储存在 -20℃可使用数周。如果所要测定的序列小于30个碱基,可将标记缓冲液稀释15倍,同时,模板DNA 要高于0.5pmol。由于DNA 模板量的不足,会降低标记反应的程度,即只标记引物后的几个核苷酸。
(2)用冰预冷的TE缓冲液按1:8稀释T7 DNA 聚合酶。酶液稀释后应立即使用,置于冰浴不宜超过60分钟。不得使用标记混合物、DTT溶液或非缓冲液类溶液稀释酶液。
(3)在已退火的引物 -模板混合物中,依次加入:
0.1mol/L DTT 1.0ml
标记混合物 2.0ml
[a-35P]dATP或[a-35S]dATP 0.5ml
已稀释好的T7 DNA 聚合酶 2.0ml
混合充分(注意不得产生气泡),置于室温5-10分钟。
如果在电泳时碰到带压缩问题,则dITP 混合物的效果优于dGTP混合物,dITP混合物的使用方法与dGTP混合物的使用是相似的。
[注意] 1、稀释时应将所有的溶液预先混合完全后再加入酶液。
2、在第(3)步的过程中,如果反应中有几次冷却,保温的时间过长或温度太高的话,则会使测序反应短于100个碱基。
3、链终止反应:
(1)取4支eppendorf管分别标上G,A,T,C。
(2)每个管分别加入2.5ml G,A,T,C链末端终止混合物,立即盖上盖子以防液体蒸发(本反应最好在标记反应前做好)。dGTP和dITP混合物依据各自需要 选用。
(3)将eppendorf管预热至37℃ 1分钟。
(4)待标记反应完毕后,取3.5ml标记反应液至上述4支eppendorf管中,稍微离心一下,并将上述四管置于37℃保温。注意在每一次反应中,均应使用新的吸液头,以免交叉污染。
(5)继续保温3-5分钟(若使用dITP时,保温时间可延长至30分钟,对反应产物没有任何影响。
(6)在上述四管中各加入4ml终止缓冲液。混合均匀后,置于冰浴中。本样品即可上样电泳。用35S标记反应的样品可置于-20℃保存一周,此时样品只有极少量的降解。而32P标记的则需在当天电泳以免降解。
(四)测序凝胶板的制备及电泳 参考第一节测序凝胶板的制备及电泳部分。样品加热至75-80℃ 2分钟,立即上样,每个泳道的使用量为2-3ml
(五)测序凝胶板的干燥及放射自显影。
电泳完毕后,经32P或35S标记的产物可用对b-射线敏感的X-光胶片放射自显影检测。
1、取下电泳的玻璃板,去除两边的压条,用小的薄钢片撬开玻璃板。若玻璃板用粘合硅烷处理过,凝胶会紧紧地粘合于玻璃板上,则凝胶与玻璃板一起进行下面步骤的处理。如果玻璃板没有经过粘合硅烷的处理,则可用3MM大滤纸贴在有凝胶的玻璃板上,小心地揭下凝胶,准备下一步的处理。
2、去除尿素,将含有凝胶的玻璃板或滤纸浸于含有2升10%冰醋酸的大号显液盆中,轻轻振荡15分钟,去除尿素。
3、干胶:含有凝胶的玻璃板可用电吹风吹干,而含有凝胶的滤纸则可用专用的干胶设备如真空加热干胶仪抽干。
4、已经干燥的凝胶即可进行放射自显影。在玻璃板含有凝胶的这一面加上X-光片后,用另一块相似大小的玻璃板夹住,然后用夹子固定,置于暗盒中曝光。而含有凝胶的滤纸则可置于X-光曝光盒中放射自显影。一般32P曝光过夜即可,而35S为2-3天。
5、曝光完毕后的X-光片即可冲洗,获得序列信息。将X光片置于水中先湿润,然后置于显影液中显影,直至条带清晰显示为止。X-光片用水淋洗片刻后置于定影液中定影15分钟以上。取出X-光片干燥并读出序列。
[ 注意] 1、去除尿素是非常重要的,如果有残存的尿素,则在干胶过程中会使凝胶很粘,而无法进行放射自显影。可以用10%冰醋酸洗二次,第一次15分钟,第二次10分钟。如果胶浓度高于12%,则有可能在干胶过程中会产生皱纹,防止的方法有二种:(1)冰醋酸溶液中加入1%-2%的甘油;(2)不干燥胶,直接在冰箱中以冰冻状态曝光,应注意的是在使用这个方法时,要在凝胶和X-光片之间加一层薄的塑料薄膜。一般来说都使用第一种方法。
2、凝胶一定要充分干燥方可进行曝光,否则X-光片会粘在胶的表面,致使以后的操作困难。
3、曝光时间应根据所使用的同位素而定,如果同位素已过半衰期则应相应延长曝光的时间。