【心得】我三年来关于AFLP的科研体会,服务大家(原创)
丁香园论坛
2309
各位战友,如果你是奋战在AFLP的战场上,我想说的是无论何时你一定得战胜平时科研中得难以名状得痛苦,甚至是老师和同学不以理解得寂寞。
AFLP的确是种技术难度很高得分子标记,从酶切到银染,步步艰辛。往往分子标记大家都觉得以PCR为基础,不至于太难,其实做出来AFLP,尤其是做出好的结果真不是件容易的事儿。
我曾经在研究生第一年没有任何结果,建立方法和平台都不知道从哪里下手,身边没有一个可以帮助我的人。我是强烈的推荐和感谢DXY,战友教会了我太多的东西,恨不得在毕业论文里致谢了。
我把我摸索出来的实验体系公布出来,献给帮助我成长的DXY:
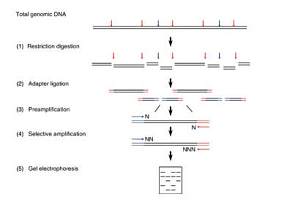
1 DNA提取采用改进的CTAB法
2 限制性消化与接头连接
2..1酶切反应体系为:
DNA(100ng/μl) 2.5μl
MseI(12U/μl) 0.25μl
EcoRI (10u/μl) 0.25μl
Buffer 4μl
BSA 0.2μl
ddH2O 12.7μl
2.2 酶切反应条件为:
37℃酶切 3h
72℃ 10min
2.3 连接体系为:
酶切完成的DNA样品 5μl
MA(25μmol/L) 1.0μl
EA(5μmol/L) 0.5μl
ATP(10mmol/L) 0.25μl
T4 DNA连接酶(3U/μl) 0.15μl
Buffer 1.0μl
ddH2O 2.1μl
2.4 连接反应条件为:
16℃ 连接过夜
3 预扩增
3 1预扩增反应体系为:
连接完成的DNA样品 1.0μl
M00(50ng/μl) 0.6μl
E00(50ng/μl) 0.6μl
10×PCR Buffer 2.0μl
Taq酶(5U/μl) 0.2μl
dNTP(10m mol/L) 0.4μl
ddH2O 15.2μl
混合后加1滴石蜡油,进行预扩增。
3.2 预扩增应条件为:
Step 1: 72℃ 5min
Step 2: 95℃ 30s
Step 3: 56℃ 30s
Step 4: 72℃ 2min
Step 5: Step 4 to Step 2 20个循环
Step 5: 72℃ 5min
Step 6: 4℃ 保温
扩增在PCR热循环仪上进行,预扩增。
扩增产物在1.5%琼脂糖检测,-20℃保存备用。
2.5 选择性扩增
2.5.1选择性扩增反应体系为:
预扩增混合液 5.0μl
MseI引物(50ng/μl) 0.8μl
EcoRI引物(50ng/μl) 0.4μl
10×PCR Buffer 2.0μl
Taq酶(2u/μl) 0.4μl
dNTP(10mmol/L) 0.4μl
ddH2O 11.0μl
混合后加1滴石蜡油,进行选择性扩增
2.5.2选择性扩增反应条件为
Step 1: 95℃ 2min;
Step 2: 95℃ 30s
Step 3: 65℃ 30s(每cycle降低0.7℃)
Step 4: 72℃ 1min
Step 5: Step 4 to Step 2: 13个循环
Step 6: 95℃ 30s
Step 7: 56℃ 30s
Step 8: 72℃ 1min
Step 9: Step 6 to Step 8: 24个循环
Step 10: 72℃ 5min
Step 11: 4℃ 保温
扩增产物1.5%琼脂糖凝胶电泳检测
-20℃保存备用
2.6.1 琼脂糖凝胶的制备
(1)取0.5×TBE缓冲液70ml,加入琼脂糖0.7克,配置成1.0%琼脂糖凝胶,微波炉中加热至沸腾;
(2)使凝胶溶液冷却至50-60℃,加入溴化乙锭,终浓度为0.5 µg/ml,轻轻旋转以充分的混匀凝胶溶液;
(3)将尚未凝结的温热的琼脂糖凝胶倒入胶膜中,排除气泡,用一个合适的梳子形成符合要求的加样孔,倒入模具;
(4)在凝胶完全凝固之后,室温下30~40 min,移去挡板和梳子,将凝胶安放倒电泳槽内,加入电泳缓冲液,没过凝胶约1mm。
2.6.2 DNA琼脂糖凝胶电泳的检测
(1)混合样品和0.2倍体积6×载样缓冲液,加样;
(2)用1~5V/cm的电压,待溴芬蓝和二甲苯氰FF迁移到适当距离后停止电泳;
(3)在紫外检测仪下检测扩增产物,记录电泳结果并照相。
2.7 变性聚丙烯酰胺凝胶电泳检测
2.7.1 测序凝胶板的制备
玻璃板的处理:银染测序的玻璃板一定要非常清洁。实验过程中,先用温水和去污剂洗涤,再用去离子水冲洗玻璃板,除去残留的去污剂,最后用乙醇清洗玻璃板。玻璃板上遗留的去污剂微膜可能导致凝胶染色时背景偏高。每次铺胶前均需分别严格处理长玻璃板和短玻璃板。
(1)长玻璃板的处理
a) 用浸透亲和硅烷溶液(1ml左右)的镜头纸擦拭清洗过的长玻璃板。长玻璃板经粘合溶液处理可将凝胶化学交联于玻璃板上;
b) 五分钟后用吸水棉纸沾95%乙醇洗长玻璃板3次,以除去多余Sigmacote溶液。
(2)短玻璃板的处理
a) 更换手套,避免与亲和硅交叉污染;
b) 用浸透剥离硅烷溶液(1.5ml)的镜头纸擦拭清洗过并已经自然干燥的整个玻璃板板面;
c) 五分钟后,用干净纸头单向擦玻璃板,然后略用力沿垂直方向擦拭。重复三次,每次均须换用干净的纸,以除去多余的粘合溶液;
d) 实验中根据温度等实际情况调整剥离硅烷的涂量。这一步对于在银染操作过程中防止凝胶撕裂至关重要。
(3)测序板处理的注意事项
a) 在单向擦玻璃板时不要过度用力,否则会带走过多粘合硅烷,使凝胶不能很好地粘附;
b) 一定要更换手套,防止粘染粘合硅烷,否则将导致凝胶撕裂;
c) 用过的凝胶可在水中浸泡后用剃须刀片或塑料刮刀刮去。玻璃板须用去污剂完全清洗。或者凝胶用10% NaOH浸泡30~60min后除去;
d) 为防止交叉污染, 用于清洗短玻璃板的工具必须与清洗长玻璃板的工具分开, 如果出现交叉污染, 以后制备的凝胶可能撕裂或变得松驰。
2.7.2 凝胶的制备:
(1) 固定玻璃板
用0.4mm厚的边条置于长玻璃板左右两侧,将短玻璃板压于其上。在长玻璃板的一侧插入鲨鱼齿梳平的一面边缘,用夹子固定住。
(2)制备测序凝胶(6%)
先用适量双蒸水溶解尿素,再加入Acr&Bis和10×TBE缓冲液,再用双蒸水调终体积至99.2ml,并用0.45mm的滤膜过滤,然后再加过硫酸铵和TEMED。
表2-1 聚丙烯酰胺凝胶浓度表
Table 2-1 table of the polyacrylamide gel strength
凝胶终浓度(%)
3 4 5 6 8 12 16 18
尿素(g) 42.0 42.0 42.0 42.0 42.0 42.0 42.0 42.0
Acr&Bis(ml) 7.5 10.0 12.0 14.5 20.0 30.0 40.0 50.0
10×TBE缓冲液(ml) 10.0 10.0 10.0 10.0 10.0 10.0 10.0 10.0
双蒸水(ml) 47.5 45.0 43.0 40.5 35.0 25.0 15.0 5.0
10%过硫酸铵(ml) 0.8 0.8 0.8 0.8 0.8 0.8 0.7 0.7
TEMED(ml) 87 80 80 80 80 70 47 40
(3)灌制胶板
将凝胶沿着压条边缘缓慢地倒入玻璃板的槽中,倒完后,静止放置使之聚合完全。一般在胶灌制后4-6分钟,即开始聚合,如果聚合不好,则应使用高浓度的TEMED和过硫酸铵。
(4)凝胶制备过程中的注意事项
a) 使用夹子固定玻璃板时,最好夹子的力量稍大一些,防止因力量不足使灌胶的过程中出现漏胶液现象。
b) 溶解尿素时不必加热。如果确需加热则应等溶液完全冷却后,方可加入TEMED和过硫酸铵。
c) 灌制凝胶的过程中要严防产生气泡,否则影响测序的结果。
2.7.3 预电泳
(1)当凝胶聚合完全后,拨出鲨鱼齿梳,将该梳子反过来,把有齿的一头插入凝胶中,形成加样孔。
(2)立即将胶板固定在测序凝胶槽中,一般测序凝胶槽的上下槽是分开的,因而只有在固定好凝胶板后,方能加入TBE缓冲液。
(3)稀释10×TBE缓冲液至1×TBE,将该缓冲液加入上下二个电泳槽中,去除产生的气泡,接上电源准备预电泳。
(4)先将水浴加热至55℃后进行预电泳。
(5)按30V/cm的电压预电泳20-30分钟(恒功率80-90W,约30min)。预电泳的过程是去除凝胶的杂质离子,同时使凝胶板达到所需的温度。高温电泳可防止GC丰富区形成的发夹状结构,影响测序的结果。
(6)预电泳的注意事项
a) 用鲨鱼齿梳制作加样孔时,应注意将齿尖插入胶中0.5mm左右,千万注意不能使加样孔渗漏,否则得不到正确的结果;
b) 应时刻注意上面电泳槽中的缓冲液是否渗漏,否则极易造成短路而损坏电泳仪;
c) 厚度小于0.4mm的胶可能导致信号太弱。
2.7.4 上样及电泳
(1)关闭电泳仪,用移液枪吸缓冲液清洗样品孔,去除在预电泳时扩散出来的尿素。
(2)PCR扩增产物:甲酰胺上样液(98%甲酰胺,10mmEDTA,0.25%溴酚兰)8:3混合,95℃变性8min,然后立即冰浴。如果样品长时间不用,则应重新处理。加样时不必吸去上层覆盖的矿物油,但要小心地吸取矿物油下的蓝色样品。AFLP选择性扩增产物95℃变性8min,然后立即冰浴。
(3)立即用毛细管进样器吸取样品,加入样品孔中。加样完毕后,立即电泳。开始可用30V/cm进行电泳,5分钟后可提高至40-60V/cm,并保持恒压状态。一般来说,一个55cm长,0.4mm厚的凝胶板,在2500V恒压状态下电泳2小时即可走到底部,同时在电泳过程中,电流可稳定地从28mA降至25mA。
(4)上样及电泳的注意事项
a)上样电泳时,一定要注意凝胶板的温度是否达到55℃左右,如果还没有达到,则应等温度达到后才能上样电泳;
b)一般来说电泳时,不宜使用太高的电压,因为太高的电压会使凝胶的分辨率降低,并且使带扩散。电泳中可进行恒功率电泳。
2.7.5 测序凝胶的银染
染色过程要求凝胶浸在塑料盘中。因而至少使用两个盘子,大小与玻璃板类似。在盘中加入新鲜溶液之前须用高质量的水洗涤盘子。
(1)电泳完毕后用一个塑料片子小心地分开两板,凝胶应该牢固地附着在长玻璃板上。
(2)固定凝胶:将凝胶(连玻璃板)放入塑料盘,用固定溶液浸没,充分振荡20分钟或至样品中染料完全消失,胶可在固定溶液中保存过夜(不振荡)。保留固定溶液,用于终止显影反应。
(3)洗胶:用超纯水振荡洗胶2次,每次8分钟。从水中取出,当转移至下一溶液时拿着胶板边沿静止10-20秒,使水流尽,再进行下一次洗涤。
(4)凝胶染色:把凝胶移至染色溶液充分摇动30分钟。
a) 显影液的配制:在已冷却至10℃的碳酸钠溶液中,加入37%甲醛(3ml)和10mg/ml的硫代硫酸钠溶液(400μl)。往染色盘中倒入预冷的反应液1升,放在一边,其余反应液仍放在冰浴中。
b) 胶从染色液中取出,放在一边,染色液回收到烧杯中,盘清洗后加入超纯水。
c) 从染色溶液中取出凝胶放入装有超纯水的盘中浸洗5-10秒。凝胶从超纯水转移到显影溶液的总时间不能长于5-10秒。浸泡时间过长则导致信号微弱或丧失信号。若浸泡时间过长,可重复用染色液浸泡处理。
d) 立刻将凝胶转移至1升(总量的一半)预冷的显影液充分振荡直至模板带开始显现或开始出现第一批条带,把凝胶移入剩下的1升显影液中继续显影2-3分钟,或直至所有条带出现。
e) 终止显色反应:弃掉显色液,加入1升终止溶液,摇床反应2-3分钟,停止显影反应,固定凝胶。
f) 洗胶:在超纯水中浸洗凝胶两次,每次2分钟,注意在本操作中戴手套拿着胶板边缘避免在胶上印上指纹。
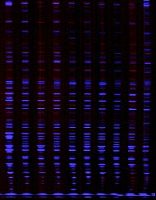
g) 干胶:将凝胶置于室温干燥或用抽气加热法干燥。在可见光灯箱或亮白,黄色背景(如纸)上观察凝胶,若需永久保存的记录, 则可用EDF胶片保留实验结果。
(5)测序产物的银染是显现序列信息的一种新方法,本系统的成败受几个因素的影响:
a) 水的质量对于染色的成功极其重要。超纯水(NANOpureR 或Milli-QR 的水)或双蒸水可获得较好的效果, 如果水中有杂质, 则低分子量条带可能无法出现;
b) 碳酸钠也非常重要。使用新鲜的,高纯度的碳酸钠,一般可获得较好的结果;
c) 染色后的洗涤步骤是非常关键的。如果凝胶洗涤时间太长,银颗粒会脱离DNA, 产生很少或没有序列信号。如果洗涤时间过长,染色步骤可以重新进行;
d) 如果凝胶厚度超过0.4mm或丙烯酰胺浓度高于4-6%,则有必要延长固定和染色的时间。如果凝胶比0.4mm薄,染色反应后的洗涤必须缩短至不超过5秒;
e) 在室温下进行所有步骤,显影反应除外。显影溶液必须预冷至10-12℃以减小背景杂色;
f) 临用前在显影溶液中加入甲醛和硫代硫酸钠。用新配的染色及显影溶液;
g) 不要重复使用任何溶液。
祝各位战友实验顺利。
AFLP的确是种技术难度很高得分子标记,从酶切到银染,步步艰辛。往往分子标记大家都觉得以PCR为基础,不至于太难,其实做出来AFLP,尤其是做出好的结果真不是件容易的事儿。
我曾经在研究生第一年没有任何结果,建立方法和平台都不知道从哪里下手,身边没有一个可以帮助我的人。我是强烈的推荐和感谢DXY,战友教会了我太多的东西,恨不得在毕业论文里致谢了。
我把我摸索出来的实验体系公布出来,献给帮助我成长的DXY:
1 DNA提取采用改进的CTAB法
2 限制性消化与接头连接
2..1酶切反应体系为:
DNA(100ng/μl) 2.5μl
MseI(12U/μl) 0.25μl
EcoRI (10u/μl) 0.25μl
Buffer 4μl
BSA 0.2μl
ddH2O 12.7μl
2.2 酶切反应条件为:
37℃酶切 3h
72℃ 10min
2.3 连接体系为:
酶切完成的DNA样品 5μl
MA(25μmol/L) 1.0μl
EA(5μmol/L) 0.5μl
ATP(10mmol/L) 0.25μl
T4 DNA连接酶(3U/μl) 0.15μl
Buffer 1.0μl
ddH2O 2.1μl
2.4 连接反应条件为:
16℃ 连接过夜
3 预扩增
3 1预扩增反应体系为:
连接完成的DNA样品 1.0μl
M00(50ng/μl) 0.6μl
E00(50ng/μl) 0.6μl
10×PCR Buffer 2.0μl
Taq酶(5U/μl) 0.2μl
dNTP(10m mol/L) 0.4μl
ddH2O 15.2μl
混合后加1滴石蜡油,进行预扩增。
3.2 预扩增应条件为:
Step 1: 72℃ 5min
Step 2: 95℃ 30s
Step 3: 56℃ 30s
Step 4: 72℃ 2min
Step 5: Step 4 to Step 2 20个循环
Step 5: 72℃ 5min
Step 6: 4℃ 保温
扩增在PCR热循环仪上进行,预扩增。
扩增产物在1.5%琼脂糖检测,-20℃保存备用。
2.5 选择性扩增
2.5.1选择性扩增反应体系为:
预扩增混合液 5.0μl
MseI引物(50ng/μl) 0.8μl
EcoRI引物(50ng/μl) 0.4μl
10×PCR Buffer 2.0μl
Taq酶(2u/μl) 0.4μl
dNTP(10mmol/L) 0.4μl
ddH2O 11.0μl
混合后加1滴石蜡油,进行选择性扩增
2.5.2选择性扩增反应条件为
Step 1: 95℃ 2min;
Step 2: 95℃ 30s
Step 3: 65℃ 30s(每cycle降低0.7℃)
Step 4: 72℃ 1min
Step 5: Step 4 to Step 2: 13个循环
Step 6: 95℃ 30s
Step 7: 56℃ 30s
Step 8: 72℃ 1min
Step 9: Step 6 to Step 8: 24个循环
Step 10: 72℃ 5min
Step 11: 4℃ 保温
扩增产物1.5%琼脂糖凝胶电泳检测
-20℃保存备用
2.6.1 琼脂糖凝胶的制备
(1)取0.5×TBE缓冲液70ml,加入琼脂糖0.7克,配置成1.0%琼脂糖凝胶,微波炉中加热至沸腾;
(2)使凝胶溶液冷却至50-60℃,加入溴化乙锭,终浓度为0.5 µg/ml,轻轻旋转以充分的混匀凝胶溶液;
(3)将尚未凝结的温热的琼脂糖凝胶倒入胶膜中,排除气泡,用一个合适的梳子形成符合要求的加样孔,倒入模具;
(4)在凝胶完全凝固之后,室温下30~40 min,移去挡板和梳子,将凝胶安放倒电泳槽内,加入电泳缓冲液,没过凝胶约1mm。
2.6.2 DNA琼脂糖凝胶电泳的检测
(1)混合样品和0.2倍体积6×载样缓冲液,加样;
(2)用1~5V/cm的电压,待溴芬蓝和二甲苯氰FF迁移到适当距离后停止电泳;
(3)在紫外检测仪下检测扩增产物,记录电泳结果并照相。
2.7 变性聚丙烯酰胺凝胶电泳检测
2.7.1 测序凝胶板的制备
玻璃板的处理:银染测序的玻璃板一定要非常清洁。实验过程中,先用温水和去污剂洗涤,再用去离子水冲洗玻璃板,除去残留的去污剂,最后用乙醇清洗玻璃板。玻璃板上遗留的去污剂微膜可能导致凝胶染色时背景偏高。每次铺胶前均需分别严格处理长玻璃板和短玻璃板。
(1)长玻璃板的处理
a) 用浸透亲和硅烷溶液(1ml左右)的镜头纸擦拭清洗过的长玻璃板。长玻璃板经粘合溶液处理可将凝胶化学交联于玻璃板上;
b) 五分钟后用吸水棉纸沾95%乙醇洗长玻璃板3次,以除去多余Sigmacote溶液。
(2)短玻璃板的处理
a) 更换手套,避免与亲和硅交叉污染;
b) 用浸透剥离硅烷溶液(1.5ml)的镜头纸擦拭清洗过并已经自然干燥的整个玻璃板板面;
c) 五分钟后,用干净纸头单向擦玻璃板,然后略用力沿垂直方向擦拭。重复三次,每次均须换用干净的纸,以除去多余的粘合溶液;
d) 实验中根据温度等实际情况调整剥离硅烷的涂量。这一步对于在银染操作过程中防止凝胶撕裂至关重要。
(3)测序板处理的注意事项
a) 在单向擦玻璃板时不要过度用力,否则会带走过多粘合硅烷,使凝胶不能很好地粘附;
b) 一定要更换手套,防止粘染粘合硅烷,否则将导致凝胶撕裂;
c) 用过的凝胶可在水中浸泡后用剃须刀片或塑料刮刀刮去。玻璃板须用去污剂完全清洗。或者凝胶用10% NaOH浸泡30~60min后除去;
d) 为防止交叉污染, 用于清洗短玻璃板的工具必须与清洗长玻璃板的工具分开, 如果出现交叉污染, 以后制备的凝胶可能撕裂或变得松驰。
2.7.2 凝胶的制备:
(1) 固定玻璃板
用0.4mm厚的边条置于长玻璃板左右两侧,将短玻璃板压于其上。在长玻璃板的一侧插入鲨鱼齿梳平的一面边缘,用夹子固定住。
(2)制备测序凝胶(6%)
先用适量双蒸水溶解尿素,再加入Acr&Bis和10×TBE缓冲液,再用双蒸水调终体积至99.2ml,并用0.45mm的滤膜过滤,然后再加过硫酸铵和TEMED。
表2-1 聚丙烯酰胺凝胶浓度表
Table 2-1 table of the polyacrylamide gel strength
凝胶终浓度(%)
3 4 5 6 8 12 16 18
尿素(g) 42.0 42.0 42.0 42.0 42.0 42.0 42.0 42.0
Acr&Bis(ml) 7.5 10.0 12.0 14.5 20.0 30.0 40.0 50.0
10×TBE缓冲液(ml) 10.0 10.0 10.0 10.0 10.0 10.0 10.0 10.0
双蒸水(ml) 47.5 45.0 43.0 40.5 35.0 25.0 15.0 5.0
10%过硫酸铵(ml) 0.8 0.8 0.8 0.8 0.8 0.8 0.7 0.7
TEMED(ml) 87 80 80 80 80 70 47 40
(3)灌制胶板
将凝胶沿着压条边缘缓慢地倒入玻璃板的槽中,倒完后,静止放置使之聚合完全。一般在胶灌制后4-6分钟,即开始聚合,如果聚合不好,则应使用高浓度的TEMED和过硫酸铵。
(4)凝胶制备过程中的注意事项
a) 使用夹子固定玻璃板时,最好夹子的力量稍大一些,防止因力量不足使灌胶的过程中出现漏胶液现象。
b) 溶解尿素时不必加热。如果确需加热则应等溶液完全冷却后,方可加入TEMED和过硫酸铵。
c) 灌制凝胶的过程中要严防产生气泡,否则影响测序的结果。
2.7.3 预电泳
(1)当凝胶聚合完全后,拨出鲨鱼齿梳,将该梳子反过来,把有齿的一头插入凝胶中,形成加样孔。
(2)立即将胶板固定在测序凝胶槽中,一般测序凝胶槽的上下槽是分开的,因而只有在固定好凝胶板后,方能加入TBE缓冲液。
(3)稀释10×TBE缓冲液至1×TBE,将该缓冲液加入上下二个电泳槽中,去除产生的气泡,接上电源准备预电泳。
(4)先将水浴加热至55℃后进行预电泳。
(5)按30V/cm的电压预电泳20-30分钟(恒功率80-90W,约30min)。预电泳的过程是去除凝胶的杂质离子,同时使凝胶板达到所需的温度。高温电泳可防止GC丰富区形成的发夹状结构,影响测序的结果。
(6)预电泳的注意事项
a) 用鲨鱼齿梳制作加样孔时,应注意将齿尖插入胶中0.5mm左右,千万注意不能使加样孔渗漏,否则得不到正确的结果;
b) 应时刻注意上面电泳槽中的缓冲液是否渗漏,否则极易造成短路而损坏电泳仪;
c) 厚度小于0.4mm的胶可能导致信号太弱。
2.7.4 上样及电泳
(1)关闭电泳仪,用移液枪吸缓冲液清洗样品孔,去除在预电泳时扩散出来的尿素。
(2)PCR扩增产物:甲酰胺上样液(98%甲酰胺,10mmEDTA,0.25%溴酚兰)8:3混合,95℃变性8min,然后立即冰浴。如果样品长时间不用,则应重新处理。加样时不必吸去上层覆盖的矿物油,但要小心地吸取矿物油下的蓝色样品。AFLP选择性扩增产物95℃变性8min,然后立即冰浴。
(3)立即用毛细管进样器吸取样品,加入样品孔中。加样完毕后,立即电泳。开始可用30V/cm进行电泳,5分钟后可提高至40-60V/cm,并保持恒压状态。一般来说,一个55cm长,0.4mm厚的凝胶板,在2500V恒压状态下电泳2小时即可走到底部,同时在电泳过程中,电流可稳定地从28mA降至25mA。
(4)上样及电泳的注意事项
a)上样电泳时,一定要注意凝胶板的温度是否达到55℃左右,如果还没有达到,则应等温度达到后才能上样电泳;
b)一般来说电泳时,不宜使用太高的电压,因为太高的电压会使凝胶的分辨率降低,并且使带扩散。电泳中可进行恒功率电泳。
2.7.5 测序凝胶的银染
染色过程要求凝胶浸在塑料盘中。因而至少使用两个盘子,大小与玻璃板类似。在盘中加入新鲜溶液之前须用高质量的水洗涤盘子。
(1)电泳完毕后用一个塑料片子小心地分开两板,凝胶应该牢固地附着在长玻璃板上。
(2)固定凝胶:将凝胶(连玻璃板)放入塑料盘,用固定溶液浸没,充分振荡20分钟或至样品中染料完全消失,胶可在固定溶液中保存过夜(不振荡)。保留固定溶液,用于终止显影反应。
(3)洗胶:用超纯水振荡洗胶2次,每次8分钟。从水中取出,当转移至下一溶液时拿着胶板边沿静止10-20秒,使水流尽,再进行下一次洗涤。
(4)凝胶染色:把凝胶移至染色溶液充分摇动30分钟。
a) 显影液的配制:在已冷却至10℃的碳酸钠溶液中,加入37%甲醛(3ml)和10mg/ml的硫代硫酸钠溶液(400μl)。往染色盘中倒入预冷的反应液1升,放在一边,其余反应液仍放在冰浴中。
b) 胶从染色液中取出,放在一边,染色液回收到烧杯中,盘清洗后加入超纯水。
c) 从染色溶液中取出凝胶放入装有超纯水的盘中浸洗5-10秒。凝胶从超纯水转移到显影溶液的总时间不能长于5-10秒。浸泡时间过长则导致信号微弱或丧失信号。若浸泡时间过长,可重复用染色液浸泡处理。
d) 立刻将凝胶转移至1升(总量的一半)预冷的显影液充分振荡直至模板带开始显现或开始出现第一批条带,把凝胶移入剩下的1升显影液中继续显影2-3分钟,或直至所有条带出现。
e) 终止显色反应:弃掉显色液,加入1升终止溶液,摇床反应2-3分钟,停止显影反应,固定凝胶。
f) 洗胶:在超纯水中浸洗凝胶两次,每次2分钟,注意在本操作中戴手套拿着胶板边缘避免在胶上印上指纹。
g) 干胶:将凝胶置于室温干燥或用抽气加热法干燥。在可见光灯箱或亮白,黄色背景(如纸)上观察凝胶,若需永久保存的记录, 则可用EDF胶片保留实验结果。
(5)测序产物的银染是显现序列信息的一种新方法,本系统的成败受几个因素的影响:
a) 水的质量对于染色的成功极其重要。超纯水(NANOpureR 或Milli-QR 的水)或双蒸水可获得较好的效果, 如果水中有杂质, 则低分子量条带可能无法出现;
b) 碳酸钠也非常重要。使用新鲜的,高纯度的碳酸钠,一般可获得较好的结果;
c) 染色后的洗涤步骤是非常关键的。如果凝胶洗涤时间太长,银颗粒会脱离DNA, 产生很少或没有序列信号。如果洗涤时间过长,染色步骤可以重新进行;
d) 如果凝胶厚度超过0.4mm或丙烯酰胺浓度高于4-6%,则有必要延长固定和染色的时间。如果凝胶比0.4mm薄,染色反应后的洗涤必须缩短至不超过5秒;
e) 在室温下进行所有步骤,显影反应除外。显影溶液必须预冷至10-12℃以减小背景杂色;
f) 临用前在显影溶液中加入甲醛和硫代硫酸钠。用新配的染色及显影溶液;
g) 不要重复使用任何溶液。
祝各位战友实验顺利。