原理
材料与仪器
Taq酶 EcoRI MseIEcoRI MseI接头 E+A引物M+C引物 T4DNALigaseE和M引物 琼脂 过硫酸胺 丙烯酰胺 尿素 硝酸银 甲酰胺 dNTPs 二甲苯青 冰醋酸 玻璃硅烷 50bpMark
电泳仪 离心机 Eppendorf管 凝胶成像仪 紫外分光光度计 37℃水浴锅 PCR仪
步骤
1. 大量提取DNA(实验方法视DNA来源而异,此处略)。
2. DNA的纯化:
(1)用0.8%琼脂糖凝胶(含EB 0.5μg/ml)电泳检测片段大小,取出其中的1/3已提取的基因组DNA进行纯化。
(2)首先用TE缓冲液补满至总体积50ul,再等体积苯酚/氯仿/异戊醇(25:24:1)、氯仿/异戊醇(24∶1)各抽提一次。
(3)离心吸上清液于Eppendorf管中,加入1/10体积的NaAC和二倍体积预冷的无水乙醇,——20℃放置2h以上。
(4)10000xg离心10min,用70%的乙醇漂洗DNA沉淀2次 ,风干后溶于30μl TE缓冲液中。
(5)紫外分光光度计检测A260、A280值并定量,再用0.8%琼脂糖凝胶(含EB 0.5μg/ml)电泳检测片段大小。
注:0.1-0.2g组织可用100ul溶液E溶,0.5g组织,溶液E可增加至300ul,此时DNA浓度大约为100ng/ul。
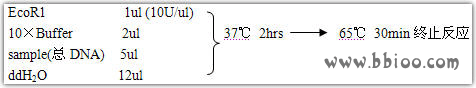
二、限制性酶切及连接
1. 在0.2ml离心管中加入:模板量约为250ng,2.5μl 10×酶切缓冲液, 2.5μl 10×T4DNA连接酶切缓冲液,5U EcoRⅠ, 5U MseⅠ,2U T4连接酶,50pmol MseⅠ接头,双蒸水补至25μl。
2. 用PCR扩增仪设定37℃过夜反应后,于65℃,20min灭酶活,——20℃保存,作为预扩增模板。
三、预扩增
1. 取3μl酶切连接产物,加入75ng E+A,75ng M+C引物,15mmol/L Mg2+,25mmol/L dNTPs,1U Tag酶,3μl 10×PCR缓冲液,加双蒸水补至30μl。
反应参数为:94℃ 90s;94℃ 30s,56℃ 1min,72℃ 1min,30循环;72℃ 10min。
2. 反应结束后,用0.8%琼脂糖凝胶(含EB 0.5μg/ml)电泳检测扩增产物,取3μl产物释稀50倍,用作选择性扩增模板。
四、选择性PCR扩增
1. 取释稀后的产物3μl,加入EcoRⅠ选择性引物、MseⅠ选择性引物各75ng,15 mmol/L Mg2+,25mmol/L dNTPs,1UTag酶,3μl 10×PCR缓冲液,加双蒸水补至30μl。
反应参数为:94℃ 90s;94℃ 30s,65℃ 1min,72℃ 1min,13循环(每循环降0.7℃);94℃ 30s, 56℃ 1min,72℃ 1min,25循环;72℃ 5min.
2. 反应结束后,用0.8%琼脂糖凝胶(含EB 0.5μg/ml)电泳检测先择性扩增产物。
五、凝胶电泳
1. 扩增产物用6%变性聚丙烯酰胺胶(厚度0.5mm)和1×TBE电泳缓冲液电泳分离)。拔出梳子,140W恒功率预电泳30分钟,温度达到47——49℃。务必使每个孔清洗出尿素。
2. 选择性扩增产物中加入等体积上样缓冲液(98%甲酰胺,10 mmol/L EDTA,0.25%二甲苯青,0.25%溴酚蓝)94℃变性5 min,结束后迅速置于冰上直到点样。
3. 每个泳道加样8μl。一开始用100W恒功率电泳约2分钟,使样品迅速集中到孔底部,再调到60W恒功率电泳,温度保持在43℃左右,待二甲苯青泳动至玻璃板2/3处,结束电泳。
六、银染
1. 固定液配制:取100ml冰醋酸到900ml去离子水或双蒸水中。
染色液:1g AgNO3,1.5ml 37% 甲醛,加去离子水至1L。
显色液:30g Na2CO3,1.5ml 37% 甲醛,2mg硫代硫酸钠,加去离子水至1L。
2. 具体操作流程
(1)电泳完毕后,将粘有凝胶的玻璃板置入用于银染的塑料盘中。
(2)固定:加入固定液,在摇床上轻微震荡30min。固定结束后,固定液保留。
(3)加入去离子水漂洗3次,每次2min。
(4)染色:将凝胶放入染色盘中,倒入染色液(4℃),在摇床上轻微震荡30min。用去离子水漂洗凝胶10秒钟后,置入显色盘中。
(5)显色:加入显色液(4℃),在摇床上轻微震荡直至条带数不再增加为止。
(6)终止:加入(2)步骤用后的固定液,来回漂几分钟。达到最好效果后,用蒸馏水漂洗几分钟。
(7)去除凝胶和玻璃板上的水珠后,放在白光灯箱上用数码相机拍照。
七、数据分析
用BIO-RAD公司的Quantity One 软件统计,再用NTSYS软件计算出遗传相似性系数,用UPGMA法进行聚类分析构建聚类图。
来源:丁香实验