【专题讨论】非放射EMSA实验技术专题讨论
丁香园论坛
1453
<center>
<strong>EMSA实验技术探讨</strong></center>
EMSA是一个比较复杂的试验技术,由于工作原因接触这个试验技术到现在也有三年多的时间了,做了大概也有几百次吧, 那倒理想的试验结果的同时也碰到了很多莫名的问题和结果,到现在为止还有没有解决的,找不到原因的一个问题,呵呵! 现和smalldonkey商量一下将我对这个实验技术的总结和一个心得写出来给大家分享,批评指正.
EMSA实验技术作为一个经典的DNA/protein,RNA/protein的检测技术,不是很多简单的实验技术可以替代的!比如曾经在我们这个论坛中有站友提到这样一个问题:
某公司的激活-和转运检测试剂盒,监测是否激活NF-KB测试剂盒原理:
NF-κB未被激活时和IκB-α形成一个复合物,分布在细胞浆中。在炎症因子、生长因子或趋化因子等可以激活NF-κB的刺激存在的情况下,IκB-α会在Ser32和Ser36被磷酸化,随后被泛素-蛋白酶体途径降解。NF-κB和IκB-α解聚后,其核定位序列被暴露,从而被转运到细胞核内促进NF-κB依赖的基因转录。通过免疫染色检测NF-κB的主要亚基p65是否被转移到细胞核内,就可以判断NF-κB是否被激活.本NF-κB激活-核转运检测试剂盒仅染色p65,不染色RelB、C-Rel、p50和p52。使用本试剂盒染色后NF-κB呈红色荧光,细胞核呈蓝色荧光.思考信号转导和检测的理论我做了这样的回答(个人意见.大家可以讨论!):
这种理论上来讲可行,但是实际操作可能拿不到你想要得结果,原因很简单,凭染色来判断是很难区分程度上的差异的,就比如做WB实验,经典的还是化学发光的方法,显色的方法只能得到信号比较强的蛋白,遇到信号比较弱的蛋白就误差很大了, 第二点,并不是所有能进核的蛋白都有结合DNA的能力,如果结合了那才是真正的参与转录的转炉银子, 有的入核后却不能和DNA结合,只能等着被降解!
其实总体而言大家做试验时都希望成功,但是EMSA试验技术的独特性和复杂性决定了,要想做成功EMSA试验,拿到阳性结果可总结为一句话: 把握整体,注意细节!
我觉的EMSA的整体性包括6大点:探针制备(设计,合成,标记,纯化,退火);个人实验设计(时间剃度浓度查阅文献等);核蛋白制备;浓度测定;EMSA操作;实验时竞争设计. 这六大点都是会直接影响到EMSA试验的成功与否! 中间就必须注意到这六大点的细节,包括操作细节,在这点上,我倒是很佩服美国佬的态度,我记得3.5年那几个美国佬给我培训的试验技术的时候就严格的按照他们的规定每一个细节操作,并养成一个实验习惯; 比如其中的一点:他们培训我们在配置溶液的时候只要是液体的东西无论什么养成一个习惯就是摇一摇,因为有些溶液静止时间长了有效的大颗粒可能下沉,要是直接取会造成不均匀的误差,到时候试验结果出了问题,很难推断操作错误的地方,养成一个习惯即使是蒸馏水也摇一摇,呵呵,倒不是说的这么夸张,但强调的是一个严格而良好的试验习惯! 呵呵,言归正传,实验技术毕竟是一门动手性很强的科学,理论还是和动手还是有一定差别的, 希望我的一些理论能给大家帮助.下面我大概的介绍一下EMSA的一些理论和自己试验的心得,以便站友学习,
另外,EMSA是一个很大的试验.我一时间也不能把所有的都罗列出来,只能等大家遇到问题,只要我有时间在线就尽力帮忙交流, 呵呵 ! 我做的最多的还是NFKB,此外还有AP-1,STAT3,STAT5,HIF等,希望能于大家交流.
简介:
EMSA ( Electrophoretic Mobility Shift Assay ) 凝胶迁移实验是一种研究DNA与蛋白质或RNA与蛋白质相互作用的常用技术。这项技术是基于DNA/蛋白质或RNA/蛋白质复合体在聚丙烯酰胺凝胶电泳(PAGE)中有不同迁移率的原理。当核转录因子与一条人工合成的特异的DNA或RNA结合后,其在PAGE中的迁移率将小于未结合核蛋白转录因子的DNA,从而检测到活化的与DNA或RNA结合的蛋白转录或调节因子。
发展:
从发展史来看,这项实验技术起初是用32P同位素标记人工合成的寡核苷酸形成探针,但是由于同位素的放射性很强,而且半衰期为14天,所以从定购到标记再到做完试验,必须14天完成,种种制约因素导致现代科技发展非同位素EMSA试验技术,这就出现了地高辛标记为探针的非放射EMSA实验技术.在实践中没多久,地高辛标记为探针的非放射EMSA实验技术就暴露了一个严重的缺陷,标记的探真纯化后灵敏度弱,导致实验结果的信号不行,最终就出现了现在的生物素标记探真的EMSA实验技术.配合化学发光技术良好的解决了灵敏度的问题,到目前为止, 生物素标记探真的EMSA实验技术广为应用!
EMSA试验成功的关键因素:1.
1.试验设计,主要是你用药品刺激细胞时,设计的药品浓度和时间剃度的问题,这个很关键,很多同学不注意这一点,最后试验确实是拿不到阳性结果,比如我一前一个朋友用药物处理SGC7901细胞,就是因为时间点设计的不好EMSA试验结果是阴性的,后来根据自己药品刺激的特性重新设计了刺激时间剃度,最终得到阳性结果! 所以这个设计很关键!!!
给一个个人的实验总结希望能帮助大家:一是受体结合类的直接刺激激活信号通路的方法,一般达到EMSA核转运高峰的时间比较短,大概控制在30min-2h之间,很多时候都是在45min和1h达到高峰,当然还有合适的浓度!!!二是是你用的是药物刺激产生受体可能时间会长一些,因为药物还有一个渗透和刺激产生细胞因子的过程.时间可以长一些,主要根据自己的药物刺激特性确定时间点.
2.核蛋白样品的制备;
制备蛋白样品的关键是:1.注意用专用的和核蛋白抽提试剂来做,才能使制备的核蛋白保持蛋白原有的天然活性和构像.2.掌握好核蛋白的浓度和纯度,尤其是浓度. 能入核的蛋白本来就不是很多,所以核蛋白的浓度往往不会很高,但是EMSA试验对核蛋白的浓度要求还是听高的! 一般要求在1ug/ul以上!有时候稍微低于这个数量级也可以,但是不能太低,否则影响结合反应拿不到结果. 这就要求核蛋白抽提尽量避免核蛋白的损失.
同时,一般制备核蛋白的材料为细胞和组织, 细胞要比组织好做的多,最重要的是用细胞得到核蛋白的纯度比组织要高很多. 而组织抽提后杂蛋白相对较多!杂蛋白多会影响试验结果不容易得到EMSA阳性结果!!
3.探针的制备:
又很多站友都在问我生物素标记到3’端还是5’端,其实我觉得最好还是两端都标记,这样才能更好的提高灵敏度, 国内的标记技术我还不是很了解,我们这边的探真都是国外定做的,还算可以,其制作程序如下: 合成寡核苷酸---标记生物素---纯化---退火合成双链.也是一个比较复杂的过程.
4.EMSA实验技术. 这一点主要是操作的细节问题! 下面会更细的谈到.
技术要点:
关于技术要点,我在这里只提一下关键的几点需要注意的细节操作,其他的照正常程序就可以了!
1. 制胶 必须是非变性PAGE凝胶,我们实验室一般用6.5%的非变性胶.制胶很重要,直接影响电泳的效果!一般控制在5分钟凝固的效果.
10X TBE 1.0ml
40% Acrylamide(40%聚丙烯酰胺) 3.3ml
50% Glycerol(50%甘油) 1.0ml
dH2O(蒸馏水) 14.8ml
TEMED(四甲基乙二氨) 20µl
脱气10min
10% AP(过硫酸氨) 120µl
总量 20.0ml
2. 一般试剂盒包括的试剂
l 10X Binding Buffer(10X 结合反应液)(-20 oC)
l Poly (dI:dC) (dI:dC)(聚核苷酸竞争物)(-20 oC)
l 6X Loading Buffer(10X 上样缓冲液)(4 oC)
l Cold oligonucleotides(非标记竞争性寡核苷酸)(-20 oC)
l Biotin-Labeled Probe(生物素标记探针)(-20 oC)
l Streptavidin-HRP(链霉亲核素-HRP)(4oC)
l 2×Blocking Buffer(2× 封闭液)(4 oC)
l 5×Washing Buffer(5× 洗涤液)(4 oC)
l Equilibration Solution(平衡液)(4 oC)
l Binding-membrane(结合反应膜)(RT)
3. 结合反应
每次结合反应需1-5µl核蛋白(根据核蛋白浓度而定),根据不同的核提取物浓度加入核提取物用量,用双蒸蒸馏水将终体积调节到15µl
(1) 结合反应体系:
10X 结合反应液 1.5µl
Poly(dI:dC)(dI:dC) 1.0µl
细胞核提取物_~N_µl
双蒸水_~N_µl
混匀室温静置20 分钟
生物素标记的探针 0.5µl
总量 15µl
混匀室温静置20分钟或以上。
(2)特异性反应确认竞争反应体系:
10X 结合反应液 1.5µl
Poly(dI:dC)(dI:dC) 1.0µl
细胞核提取物 ? µl
未标记的竞争性寡核 2.0µl
双蒸水_~N_µl
混匀室温静置20 分钟
生物素标记的探针 0.5µl
总量 15µl
混匀室温静置20分钟或以上。
其中:
探针用量: 这相当于10-50fmoles的DNA探针。我们通常是最少50fmol.
蛋白样品用量: 一般用量在2---20ug(控制在1-5µl)蛋白, 总体系15ul. 细胞核抽屉物浓度都比较低,组织的一般都比较高,因为杂蛋白较多.
poly(dI:dC)(dI:dC): 它由肌苷和胞嘧啶组成。由于其特定的结构,可抑制蛋白对标记探针的非特异结合,避免假复合物。 在凝胶迁移反应中加入poly(dI:dC)(dI:dC),可抑制粗制核抽提液中其它DNA结合蛋白结合,比如转录调节因子的非特异结合。当用纯化的蛋白作凝胶迁移反应时,不必一定加入poly(dI:dC)(dI:dC),如加入,则普通反应中所用终浓度不超过50-100ng。对核抽提液:每2-3ug核抽提液用1 ug poly(dI:dC)(dI:dC)。
未标记的竞争性寡核: 做为竞争的探真来说,其实就是没有标记的转炉银子的寡核苷酸,用量一般是正常探真的50-100倍. 再这里我引申的解释一下其他的对照的含义:
◆. 常规反应(含激活的目的转录因子的核蛋白+标记探针);
◆ 阴性对照反应(核蛋白+标记探针);
◆ 阳性对照(核蛋白+标记探针)
◆ 探针冷竞争反应(含激活的目的转录因子的核蛋白+标记探针+标记探针100倍量的未标记探针);
◆ 突变探针的冷竞争反应(含激活的目的转录因子的 核蛋白+标记探针+标记探针100倍量的未标记突变探针);
◆ Super-shift反应(含激活的目的转录因子的核蛋白+标记探针+目的转录因子的特异抗体).
4. 预电泳和电泳:0.25X TBE在冰上120V 预电泳1小时; 务必换掉预电泳的缓冲液, 用新的0.25X TB低温下150V,电泳约60分钟。 注意横压电泳的时候注意电流的数字变化,记录开始电泳和电泳结束的电流数据的变化!
5. 电转运
在0.5X TBE中390mA电转移40分钟, 注意转运膜的选择, 有一种离子加强型的转运效果比较好! 我当初选择过一般的和离子加强型的,还是离子加强型结合效果好!
6.紫外交联: 正规的应该是紫外交联仪的使用,但是很多单位没有交联仪,那就用无菌操作台上的紫外等下10cm处交联10分钟就可以了!这个数据是我做了好多次试验才摸索出来的!
条件比较成熟!可以借鉴!
7. 化学发光反应
包括Blocking Buffer 30分钟封闭, Streptavidin-HRP标记, Washing Buffer洗涤; Equilibration Solution平衡, 这些按照规定操作即可! 没有太多的细节,只是注意两点,一是期间结合膜的表面一定不能干燥!!!二是Streptavidin-HRP不能加在膜上,而是加在液体中混韵后在将膜放在液体中孵浴.
8. 化学发光图像显示
两种方法:化学发光数字成像与检测和感光胶片冲印成像
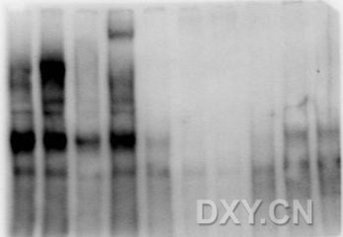
操作基本和Western 试验操作相当, 只是这个膜是一次性的,不能重复使用, 一定保存好图片!!! 由于跑得是非变性凝胶,蛋白保持天然构想和活性,所以代条很可能是一块一块的而不像WB那样很规整的一条细线,这个很正常!!!
希望大家交流讨论!
EMSA是一个比较复杂的试验技术,由于工作原因接触这个试验技术到现在也有三年多的时间了,做了大概也有几百次吧, 那倒理想的试验结果的同时也碰到了很多莫名的问题和结果,到现在为止还有没有解决的,找不到原因的一个问题,呵呵! 现和smalldonkey商量一下将我对这个实验技术的总结和一个心得写出来给大家分享,批评指正.
EMSA实验技术作为一个经典的DNA/protein,RNA/protein的检测技术,不是很多简单的实验技术可以替代的!比如曾经在我们这个论坛中有站友提到这样一个问题:
某公司的激活-和转运检测试剂盒,监测是否激活NF-KB测试剂盒原理:
NF-κB未被激活时和IκB-α形成一个复合物,分布在细胞浆中。在炎症因子、生长因子或趋化因子等可以激活NF-κB的刺激存在的情况下,IκB-α会在Ser32和Ser36被磷酸化,随后被泛素-蛋白酶体途径降解。NF-κB和IκB-α解聚后,其核定位序列被暴露,从而被转运到细胞核内促进NF-κB依赖的基因转录。通过免疫染色检测NF-κB的主要亚基p65是否被转移到细胞核内,就可以判断NF-κB是否被激活.本NF-κB激活-核转运检测试剂盒仅染色p65,不染色RelB、C-Rel、p50和p52。使用本试剂盒染色后NF-κB呈红色荧光,细胞核呈蓝色荧光.思考信号转导和检测的理论我做了这样的回答(个人意见.大家可以讨论!):
这种理论上来讲可行,但是实际操作可能拿不到你想要得结果,原因很简单,凭染色来判断是很难区分程度上的差异的,就比如做WB实验,经典的还是化学发光的方法,显色的方法只能得到信号比较强的蛋白,遇到信号比较弱的蛋白就误差很大了, 第二点,并不是所有能进核的蛋白都有结合DNA的能力,如果结合了那才是真正的参与转录的转炉银子, 有的入核后却不能和DNA结合,只能等着被降解!
其实总体而言大家做试验时都希望成功,但是EMSA试验技术的独特性和复杂性决定了,要想做成功EMSA试验,拿到阳性结果可总结为一句话: 把握整体,注意细节!
我觉的EMSA的整体性包括6大点:探针制备(设计,合成,标记,纯化,退火);个人实验设计(时间剃度浓度查阅文献等);核蛋白制备;浓度测定;EMSA操作;实验时竞争设计. 这六大点都是会直接影响到EMSA试验的成功与否! 中间就必须注意到这六大点的细节,包括操作细节,在这点上,我倒是很佩服美国佬的态度,我记得3.5年那几个美国佬给我培训的试验技术的时候就严格的按照他们的规定每一个细节操作,并养成一个实验习惯; 比如其中的一点:他们培训我们在配置溶液的时候只要是液体的东西无论什么养成一个习惯就是摇一摇,因为有些溶液静止时间长了有效的大颗粒可能下沉,要是直接取会造成不均匀的误差,到时候试验结果出了问题,很难推断操作错误的地方,养成一个习惯即使是蒸馏水也摇一摇,呵呵,倒不是说的这么夸张,但强调的是一个严格而良好的试验习惯! 呵呵,言归正传,实验技术毕竟是一门动手性很强的科学,理论还是和动手还是有一定差别的, 希望我的一些理论能给大家帮助.下面我大概的介绍一下EMSA的一些理论和自己试验的心得,以便站友学习,
另外,EMSA是一个很大的试验.我一时间也不能把所有的都罗列出来,只能等大家遇到问题,只要我有时间在线就尽力帮忙交流, 呵呵 ! 我做的最多的还是NFKB,此外还有AP-1,STAT3,STAT5,HIF等,希望能于大家交流.
简介:
EMSA ( Electrophoretic Mobility Shift Assay ) 凝胶迁移实验是一种研究DNA与蛋白质或RNA与蛋白质相互作用的常用技术。这项技术是基于DNA/蛋白质或RNA/蛋白质复合体在聚丙烯酰胺凝胶电泳(PAGE)中有不同迁移率的原理。当核转录因子与一条人工合成的特异的DNA或RNA结合后,其在PAGE中的迁移率将小于未结合核蛋白转录因子的DNA,从而检测到活化的与DNA或RNA结合的蛋白转录或调节因子。
发展:
从发展史来看,这项实验技术起初是用32P同位素标记人工合成的寡核苷酸形成探针,但是由于同位素的放射性很强,而且半衰期为14天,所以从定购到标记再到做完试验,必须14天完成,种种制约因素导致现代科技发展非同位素EMSA试验技术,这就出现了地高辛标记为探针的非放射EMSA实验技术.在实践中没多久,地高辛标记为探针的非放射EMSA实验技术就暴露了一个严重的缺陷,标记的探真纯化后灵敏度弱,导致实验结果的信号不行,最终就出现了现在的生物素标记探真的EMSA实验技术.配合化学发光技术良好的解决了灵敏度的问题,到目前为止, 生物素标记探真的EMSA实验技术广为应用!
EMSA试验成功的关键因素:1.
1.试验设计,主要是你用药品刺激细胞时,设计的药品浓度和时间剃度的问题,这个很关键,很多同学不注意这一点,最后试验确实是拿不到阳性结果,比如我一前一个朋友用药物处理SGC7901细胞,就是因为时间点设计的不好EMSA试验结果是阴性的,后来根据自己药品刺激的特性重新设计了刺激时间剃度,最终得到阳性结果! 所以这个设计很关键!!!
给一个个人的实验总结希望能帮助大家:一是受体结合类的直接刺激激活信号通路的方法,一般达到EMSA核转运高峰的时间比较短,大概控制在30min-2h之间,很多时候都是在45min和1h达到高峰,当然还有合适的浓度!!!二是是你用的是药物刺激产生受体可能时间会长一些,因为药物还有一个渗透和刺激产生细胞因子的过程.时间可以长一些,主要根据自己的药物刺激特性确定时间点.
2.核蛋白样品的制备;
制备蛋白样品的关键是:1.注意用专用的和核蛋白抽提试剂来做,才能使制备的核蛋白保持蛋白原有的天然活性和构像.2.掌握好核蛋白的浓度和纯度,尤其是浓度. 能入核的蛋白本来就不是很多,所以核蛋白的浓度往往不会很高,但是EMSA试验对核蛋白的浓度要求还是听高的! 一般要求在1ug/ul以上!有时候稍微低于这个数量级也可以,但是不能太低,否则影响结合反应拿不到结果. 这就要求核蛋白抽提尽量避免核蛋白的损失.
同时,一般制备核蛋白的材料为细胞和组织, 细胞要比组织好做的多,最重要的是用细胞得到核蛋白的纯度比组织要高很多. 而组织抽提后杂蛋白相对较多!杂蛋白多会影响试验结果不容易得到EMSA阳性结果!!
3.探针的制备:
又很多站友都在问我生物素标记到3’端还是5’端,其实我觉得最好还是两端都标记,这样才能更好的提高灵敏度, 国内的标记技术我还不是很了解,我们这边的探真都是国外定做的,还算可以,其制作程序如下: 合成寡核苷酸---标记生物素---纯化---退火合成双链.也是一个比较复杂的过程.
4.EMSA实验技术. 这一点主要是操作的细节问题! 下面会更细的谈到.
技术要点:
关于技术要点,我在这里只提一下关键的几点需要注意的细节操作,其他的照正常程序就可以了!
1. 制胶 必须是非变性PAGE凝胶,我们实验室一般用6.5%的非变性胶.制胶很重要,直接影响电泳的效果!一般控制在5分钟凝固的效果.
10X TBE 1.0ml
40% Acrylamide(40%聚丙烯酰胺) 3.3ml
50% Glycerol(50%甘油) 1.0ml
dH2O(蒸馏水) 14.8ml
TEMED(四甲基乙二氨) 20µl
脱气10min
10% AP(过硫酸氨) 120µl
总量 20.0ml
2. 一般试剂盒包括的试剂
l 10X Binding Buffer(10X 结合反应液)(-20 oC)
l Poly (dI:dC) (dI:dC)(聚核苷酸竞争物)(-20 oC)
l 6X Loading Buffer(10X 上样缓冲液)(4 oC)
l Cold oligonucleotides(非标记竞争性寡核苷酸)(-20 oC)
l Biotin-Labeled Probe(生物素标记探针)(-20 oC)
l Streptavidin-HRP(链霉亲核素-HRP)(4oC)
l 2×Blocking Buffer(2× 封闭液)(4 oC)
l 5×Washing Buffer(5× 洗涤液)(4 oC)
l Equilibration Solution(平衡液)(4 oC)
l Binding-membrane(结合反应膜)(RT)
3. 结合反应
每次结合反应需1-5µl核蛋白(根据核蛋白浓度而定),根据不同的核提取物浓度加入核提取物用量,用双蒸蒸馏水将终体积调节到15µl
(1) 结合反应体系:
10X 结合反应液 1.5µl
Poly(dI:dC)(dI:dC) 1.0µl
细胞核提取物_~N_µl
双蒸水_~N_µl
混匀室温静置20 分钟
生物素标记的探针 0.5µl
总量 15µl
混匀室温静置20分钟或以上。
(2)特异性反应确认竞争反应体系:
10X 结合反应液 1.5µl
Poly(dI:dC)(dI:dC) 1.0µl
细胞核提取物 ? µl
未标记的竞争性寡核 2.0µl
双蒸水_~N_µl
混匀室温静置20 分钟
生物素标记的探针 0.5µl
总量 15µl
混匀室温静置20分钟或以上。
其中:
探针用量: 这相当于10-50fmoles的DNA探针。我们通常是最少50fmol.
蛋白样品用量: 一般用量在2---20ug(控制在1-5µl)蛋白, 总体系15ul. 细胞核抽屉物浓度都比较低,组织的一般都比较高,因为杂蛋白较多.
poly(dI:dC)(dI:dC): 它由肌苷和胞嘧啶组成。由于其特定的结构,可抑制蛋白对标记探针的非特异结合,避免假复合物。 在凝胶迁移反应中加入poly(dI:dC)(dI:dC),可抑制粗制核抽提液中其它DNA结合蛋白结合,比如转录调节因子的非特异结合。当用纯化的蛋白作凝胶迁移反应时,不必一定加入poly(dI:dC)(dI:dC),如加入,则普通反应中所用终浓度不超过50-100ng。对核抽提液:每2-3ug核抽提液用1 ug poly(dI:dC)(dI:dC)。
未标记的竞争性寡核: 做为竞争的探真来说,其实就是没有标记的转炉银子的寡核苷酸,用量一般是正常探真的50-100倍. 再这里我引申的解释一下其他的对照的含义:
◆. 常规反应(含激活的目的转录因子的核蛋白+标记探针);
◆ 阴性对照反应(核蛋白+标记探针);
◆ 阳性对照(核蛋白+标记探针)
◆ 探针冷竞争反应(含激活的目的转录因子的核蛋白+标记探针+标记探针100倍量的未标记探针);
◆ 突变探针的冷竞争反应(含激活的目的转录因子的 核蛋白+标记探针+标记探针100倍量的未标记突变探针);
◆ Super-shift反应(含激活的目的转录因子的核蛋白+标记探针+目的转录因子的特异抗体).
4. 预电泳和电泳:0.25X TBE在冰上120V 预电泳1小时; 务必换掉预电泳的缓冲液, 用新的0.25X TB低温下150V,电泳约60分钟。 注意横压电泳的时候注意电流的数字变化,记录开始电泳和电泳结束的电流数据的变化!
5. 电转运
在0.5X TBE中390mA电转移40分钟, 注意转运膜的选择, 有一种离子加强型的转运效果比较好! 我当初选择过一般的和离子加强型的,还是离子加强型结合效果好!
6.紫外交联: 正规的应该是紫外交联仪的使用,但是很多单位没有交联仪,那就用无菌操作台上的紫外等下10cm处交联10分钟就可以了!这个数据是我做了好多次试验才摸索出来的!
条件比较成熟!可以借鉴!
7. 化学发光反应
包括Blocking Buffer 30分钟封闭, Streptavidin-HRP标记, Washing Buffer洗涤; Equilibration Solution平衡, 这些按照规定操作即可! 没有太多的细节,只是注意两点,一是期间结合膜的表面一定不能干燥!!!二是Streptavidin-HRP不能加在膜上,而是加在液体中混韵后在将膜放在液体中孵浴.
8. 化学发光图像显示
两种方法:化学发光数字成像与检测和感光胶片冲印成像
操作基本和Western 试验操作相当, 只是这个膜是一次性的,不能重复使用, 一定保存好图片!!! 由于跑得是非变性凝胶,蛋白保持天然构想和活性,所以代条很可能是一块一块的而不像WB那样很规整的一条细线,这个很正常!!!
希望大家交流讨论!