【原创】表达载体的选择及构建方法
丁香园论坛
24718
<center>
</center>
<center>
<strong>表达载体的构建方法及步骤</strong></center>
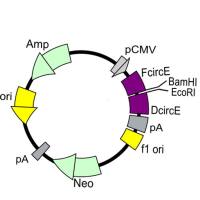
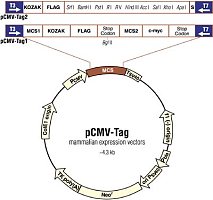
一、载体的选择及如何阅读质粒图谱
目前,载体主要有病毒和非病毒两大类,其中质粒 DNA 是一种新的非病毒转基因载体。
一个合格质粒的组成要素:
(1)复制起始位点 Ori 即控制复制起始的位点。原核生物 DNA 分子中只有一个复制起始点。而
真核生物 DNA 分子有多个复制起始位点。
(2)抗生素抗性基因可以便于加以检测,如 Amp+ ,Kan+
(3)多克隆位点 MCS 克隆携带外源基因片段
(4) P/E 启动子/增强子
(5)Terms 终止信号
(6)加 poly(A)信号可以起到稳定 mRNA 作用
选择载体主要依据构建的目的,同时要考虑载体中应有合适的限制酶切位点。如果构建的目
的是要表达一个特定的基因,则要选择合适的表达载体。
载体选择主要考虑下述3点:
【1】构建 DNA 重组体的目的,克隆扩增/基因表达,选择合适的克隆载体/表达载体。
【2】.载体的类型:
(1)克隆载体的克隆能力-据克隆片段大小(大选大,小选小)。如<10kb 选质粒。
(2)表达载体据受体细胞类型-原核/真核/穿梭,E.coli/哺乳类细胞表达载体。
(3)对原核表达载体应该注意:选择合适的启动子及相应的受体菌,用于表达真核蛋白质时注意克服4个困难和阅读框错位;表达天然蛋白质或融合蛋白作为相应载体的参考。
【3】载体 MCS 中的酶切位点数与组成方向因载体不同而异,适应目的基因与载体易于链接,不能产生阅读框架错位。
综上所述,选用质粒(最常用)做载体的5点要求:
(1)选分子量小的质粒,即小载体(1-1.5kb)→不易损坏,在细菌里面拷贝数也多(也有大载
体);
(2)一般使用松弛型质粒在细菌里扩增不受约束,一般 10个以上的拷贝,而严谨型质粒<10个。
(3)必需具备一个以上的酶切位点,有选择的余地;
(4)必需有易检测的标记,多是抗生素的抗性基因,不特指多位 Ampr(试一试)。
(5)满足自己的实验需求,是否需要包装病毒,是否需要加入荧光标记,是否需要加入标签蛋白,是否需要真核抗性(如Puro、G418)等等。
无论选用哪种载体,首先都要获得载体分子,然后采用适当的限制酶将载体 DNA 进行切割,获得线性载体分子,以便于与目的基因片段进行连接。
如何阅读质粒图谱
第一步:首先看 Ori 的位置,了解质粒的类型(原核/真核/穿梭质粒)
第二步:再看筛选标记,如抗性,决定使用什么筛选标记。
(1)Ampr 水解β-内酰胺环,解除氨苄的毒性。
(2)tetr 可以阻止四环素进入细胞。
(3)camr 生成氯霉素羟乙酰基衍生物,使之失去毒性。
(4)neor(kanr)氨基糖苷磷酸转移酶使 G418(长那霉素衍生物)失活
(5)hygr 使潮霉素β失活。
第三步:看多克隆位点(MCS)。它具有多个限制酶的单一切点。便于外源基因的插入。如果在这些酶切位点以外有外源基因的插入,会导致某种标志基因的失活,而便于筛选。决定能不能放目的基因以及如何放置目的基因。
第四步:再看外源 DNA 插入片段大小。质粒一般只能容纳小于10Kb 的外源 DNA 片段。一般来说,外源 DNA 片段越长,越难插入,越不稳定,转化效率越低。
第五步:是否含有表达系统元件,即启动子-核糖体结合位点-克隆位点-转录终止信号。这是用来区别克隆载体与表达载体。克隆载体中加入一些与表达调控有关的元件即成为表达载体。选用那种载体,还是要以实验目的为准绳。
第六步:启动子-核糖体结合位点-克隆位点-转录终止信号
(1)启动子-促进 DNA 转录的 DNA 序列,这个 DNA 区域常在基因或操纵子编码序列的上游,是 DNA 分子上可以与 RNApol 特异性结合并使之开始转录的部位,但启动子本身不被转录。
(2)增强子/沉默子-为真核基因组(包括真核病毒基因组)中的一种具有增强邻近基因转录过程的调控顺序。其作用与增强子所在的位置或方向无关。即在所调控基因上游或下游均可发挥作用。/沉默子-负增强子,负调控序列。
(3)核糖体结合位点/起始密码/SD 序列(Rbs/AGU/SDs):mRNA 有核糖体的两个结合位点,对于原核而言是 AUG(起始密码)和 SD 序列。
(4)转录终止序列(终止子)/翻译终止密码子:结构基因的最后一个外显子中有一个 AATAAA的保守序列,此位点 down-stream 有一段 GT 或 T 富丰区,这2部分共同构成 poly(A)加尾信号。结构基因的最后一个外显子中有一个 AATAAA 的保守序列,此位点 down-stream 有一段GT 或 T 富丰区,这2部分共同构成 poly(A)加尾信号。
质粒图谱上有的箭头顺时针有的箭头逆时针,那其实是代表两条 DNA 链,即质粒是环状双链DNA,它的启动子等在其中一条链上,而它的抗性基因在另一条链上 .根据表达宿主不同,构建时所选择的载体也会不同。
二、目的基因的获得
一般来说,目的基因的获得有三种途径:
调取基因:根据目的基因的序列,设计引物从含有目的基因的cDNA中通过PCR的方法调取目的基因,链接到克隆载体挑取单克隆进行测序,以获得想要的基因片段,这种方法相对成本较低,但是调取到的基因往往含有突变,还有不同基因的表达丰度不同,转录本比较复杂,或是基因片段很长,这些情况都很难调取到目的基因。
全基因合成:根据目的基因的DNA序列,直接设计合成目的基因。此方法准确性高,相对成本会高一些,个人操作比较困难,需要专业的合成公司完成。优点是可以合成难调取及人工改造的任何基因序列,同时可以进行密码子优化,提高目的基因在宿主内的表达量。
三、克隆构建
目前,克隆构建的方法多种多样,除了应用广泛的酶切链接以外,现在还有很多不依赖酶切位点的克隆构建方式。下面具体说一下双酶切方法构建载体的步骤。
实验材料
实验试剂
(2)X基因慢病毒载体的构建
X基因基因由Transheep全基因合成,构建于载体PUC57中。
PUC57-X基因 EcoRI/BamHI酶切结果:
<center> <img height="239" src="http://assets.dxycdn.com/third-party/xheditor/xheditor_skin/blank.gif" width="113" /></center> <center> <img height="131" src="http://assets.dxycdn.com/third-party/xheditor/xheditor_skin/blank.gif" width="484" /></center> 酶切完成后进行胶回收
2. 载体用pCDNA3.1双酶切,酶切体系如下。
20ul酶切体系 37度3小时
4ul pCDNA3.1载体(500 ng/ul)
1ul BamHI
1ul EcoRI
2ul 10×buffer
12 ul H2O
酶切完成后胶回收(见附录)
<center> <img height="205" src="http://assets.dxycdn.com/third-party/xheditor/xheditor_skin/blank.gif" width="73" /><img height="110" src="http://assets.dxycdn.com/third-party/xheditor/xheditor_skin/blank.gif" width="596" /></center> 处理好的目的片段与载体连接反应体系:
6ul PCR 酶切回收片段(约50ng/ul)
1ul 酶切好的载体(约50ng/ul)
2ul ligase buffer
1ul T4ligase
10ul H2O
以上连接液在16℃过夜。
转化 (感受态细胞: DH5a),具体步骤见附录转化部分。
抗性: Amp; 37℃,培养过夜
转化后X基因分别平板挑菌, 37℃ 250转/分钟摇菌14小时,PCR鉴定后,将阳性菌液送上海权阳生物技术有限公司测序。
X基因慢病毒载体单克隆菌落PCR鉴定(使用载体通用引物,PCR条带大小应比实际大200bp左右):
<center> <img height="173" src="http://assets.dxycdn.com/third-party/xheditor/xheditor_skin/blank.gif" width="158" /></center>
一、载体的选择及如何阅读质粒图谱
目前,载体主要有病毒和非病毒两大类,其中质粒 DNA 是一种新的非病毒转基因载体。
一个合格质粒的组成要素:
(1)复制起始位点 Ori 即控制复制起始的位点。原核生物 DNA 分子中只有一个复制起始点。而
真核生物 DNA 分子有多个复制起始位点。
(2)抗生素抗性基因可以便于加以检测,如 Amp+ ,Kan+
(3)多克隆位点 MCS 克隆携带外源基因片段
(4) P/E 启动子/增强子
(5)Terms 终止信号
(6)加 poly(A)信号可以起到稳定 mRNA 作用
选择载体主要依据构建的目的,同时要考虑载体中应有合适的限制酶切位点。如果构建的目
的是要表达一个特定的基因,则要选择合适的表达载体。
载体选择主要考虑下述3点:
【1】构建 DNA 重组体的目的,克隆扩增/基因表达,选择合适的克隆载体/表达载体。
【2】.载体的类型:
(1)克隆载体的克隆能力-据克隆片段大小(大选大,小选小)。如<10kb 选质粒。
(2)表达载体据受体细胞类型-原核/真核/穿梭,E.coli/哺乳类细胞表达载体。
(3)对原核表达载体应该注意:选择合适的启动子及相应的受体菌,用于表达真核蛋白质时注意克服4个困难和阅读框错位;表达天然蛋白质或融合蛋白作为相应载体的参考。
【3】载体 MCS 中的酶切位点数与组成方向因载体不同而异,适应目的基因与载体易于链接,不能产生阅读框架错位。
综上所述,选用质粒(最常用)做载体的5点要求:
(1)选分子量小的质粒,即小载体(1-1.5kb)→不易损坏,在细菌里面拷贝数也多(也有大载
体);
(2)一般使用松弛型质粒在细菌里扩增不受约束,一般 10个以上的拷贝,而严谨型质粒<10个。
(3)必需具备一个以上的酶切位点,有选择的余地;
(4)必需有易检测的标记,多是抗生素的抗性基因,不特指多位 Ampr(试一试)。
(5)满足自己的实验需求,是否需要包装病毒,是否需要加入荧光标记,是否需要加入标签蛋白,是否需要真核抗性(如Puro、G418)等等。
无论选用哪种载体,首先都要获得载体分子,然后采用适当的限制酶将载体 DNA 进行切割,获得线性载体分子,以便于与目的基因片段进行连接。
如何阅读质粒图谱
第一步:首先看 Ori 的位置,了解质粒的类型(原核/真核/穿梭质粒)
第二步:再看筛选标记,如抗性,决定使用什么筛选标记。
(1)Ampr 水解β-内酰胺环,解除氨苄的毒性。
(2)tetr 可以阻止四环素进入细胞。
(3)camr 生成氯霉素羟乙酰基衍生物,使之失去毒性。
(4)neor(kanr)氨基糖苷磷酸转移酶使 G418(长那霉素衍生物)失活
(5)hygr 使潮霉素β失活。
第三步:看多克隆位点(MCS)。它具有多个限制酶的单一切点。便于外源基因的插入。如果在这些酶切位点以外有外源基因的插入,会导致某种标志基因的失活,而便于筛选。决定能不能放目的基因以及如何放置目的基因。
第四步:再看外源 DNA 插入片段大小。质粒一般只能容纳小于10Kb 的外源 DNA 片段。一般来说,外源 DNA 片段越长,越难插入,越不稳定,转化效率越低。
第五步:是否含有表达系统元件,即启动子-核糖体结合位点-克隆位点-转录终止信号。这是用来区别克隆载体与表达载体。克隆载体中加入一些与表达调控有关的元件即成为表达载体。选用那种载体,还是要以实验目的为准绳。
第六步:启动子-核糖体结合位点-克隆位点-转录终止信号
(1)启动子-促进 DNA 转录的 DNA 序列,这个 DNA 区域常在基因或操纵子编码序列的上游,是 DNA 分子上可以与 RNApol 特异性结合并使之开始转录的部位,但启动子本身不被转录。
(2)增强子/沉默子-为真核基因组(包括真核病毒基因组)中的一种具有增强邻近基因转录过程的调控顺序。其作用与增强子所在的位置或方向无关。即在所调控基因上游或下游均可发挥作用。/沉默子-负增强子,负调控序列。
(3)核糖体结合位点/起始密码/SD 序列(Rbs/AGU/SDs):mRNA 有核糖体的两个结合位点,对于原核而言是 AUG(起始密码)和 SD 序列。
(4)转录终止序列(终止子)/翻译终止密码子:结构基因的最后一个外显子中有一个 AATAAA的保守序列,此位点 down-stream 有一段 GT 或 T 富丰区,这2部分共同构成 poly(A)加尾信号。结构基因的最后一个外显子中有一个 AATAAA 的保守序列,此位点 down-stream 有一段GT 或 T 富丰区,这2部分共同构成 poly(A)加尾信号。
质粒图谱上有的箭头顺时针有的箭头逆时针,那其实是代表两条 DNA 链,即质粒是环状双链DNA,它的启动子等在其中一条链上,而它的抗性基因在另一条链上 .根据表达宿主不同,构建时所选择的载体也会不同。
二、目的基因的获得
一般来说,目的基因的获得有三种途径:
调取基因:根据目的基因的序列,设计引物从含有目的基因的cDNA中通过PCR的方法调取目的基因,链接到克隆载体挑取单克隆进行测序,以获得想要的基因片段,这种方法相对成本较低,但是调取到的基因往往含有突变,还有不同基因的表达丰度不同,转录本比较复杂,或是基因片段很长,这些情况都很难调取到目的基因。
全基因合成:根据目的基因的DNA序列,直接设计合成目的基因。此方法准确性高,相对成本会高一些,个人操作比较困难,需要专业的合成公司完成。优点是可以合成难调取及人工改造的任何基因序列,同时可以进行密码子优化,提高目的基因在宿主内的表达量。
三、克隆构建
目前,克隆构建的方法多种多样,除了应用广泛的酶切链接以外,现在还有很多不依赖酶切位点的克隆构建方式。下面具体说一下双酶切方法构建载体的步骤。
实验材料
实验试剂
| 试剂名称 | 生产厂家 |
| 载体pCDNA3.1 | Transheep |
| 大肠杆菌菌株DH5α | Tiangen |
| 限制性内切酶 | Fermentas |
| T4连接酶 | Fermentas |
| 质粒DNA小,大量抽提试剂盒 | Axygen |
| 凝胶回收试剂盒 | Axygen |
| 琼脂糖 | Biowest |
| DNA ladder | Fermentas |
(2)X基因慢病毒载体的构建
X基因基因由Transheep全基因合成,构建于载体PUC57中。
PUC57-X基因 EcoRI/BamHI酶切结果:
<center> <img height="239" src="http://assets.dxycdn.com/third-party/xheditor/xheditor_skin/blank.gif" width="113" /></center> <center> <img height="131" src="http://assets.dxycdn.com/third-party/xheditor/xheditor_skin/blank.gif" width="484" /></center> 酶切完成后进行胶回收
2. 载体用pCDNA3.1双酶切,酶切体系如下。
20ul酶切体系 37度3小时
4ul pCDNA3.1载体(500 ng/ul)
1ul BamHI
1ul EcoRI
2ul 10×buffer
12 ul H2O
酶切完成后胶回收(见附录)
<center> <img height="205" src="http://assets.dxycdn.com/third-party/xheditor/xheditor_skin/blank.gif" width="73" /><img height="110" src="http://assets.dxycdn.com/third-party/xheditor/xheditor_skin/blank.gif" width="596" /></center> 处理好的目的片段与载体连接反应体系:
6ul PCR 酶切回收片段(约50ng/ul)
1ul 酶切好的载体(约50ng/ul)
2ul ligase buffer
1ul T4ligase
10ul H2O
以上连接液在16℃过夜。
转化 (感受态细胞: DH5a),具体步骤见附录转化部分。
抗性: Amp; 37℃,培养过夜
转化后X基因分别平板挑菌, 37℃ 250转/分钟摇菌14小时,PCR鉴定后,将阳性菌液送上海权阳生物技术有限公司测序。
X基因慢病毒载体单克隆菌落PCR鉴定(使用载体通用引物,PCR条带大小应比实际大200bp左右):
<center> <img height="173" src="http://assets.dxycdn.com/third-party/xheditor/xheditor_skin/blank.gif" width="158" /></center>