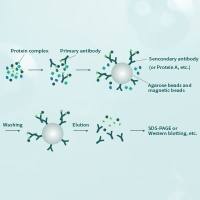
实战指南——免疫共沉淀篇(进阶)
互联网
非内源性蛋白的相互作用,主要是通过过表达来实现,通常为简化实验、提高IP效率会让外源蛋白带上标签(tagged;这也是没有相应的可用于IP的内源蛋白抗体之前唯一可行的方案),因此就tag的使用而言存在很多小技巧。
2. tandem-tagged不能用于分析钙调信号通路。tandem tag实际上从成本角度和His tag差不多,洗脱试剂价格低廉,因此可用于大规模PD之后的质谱分析,能获取最全面的相互作用的信息。然后由于其中包含钙调蛋白相互作用domain,天然会结合钙信号相关蛋白,从而干扰或掩盖靶蛋白与钙信号蛋白之间的相互作用,有一定的应用局限。当然,笔者不太清楚近年来该方法有无改进,但正是由于引入钙调domain才实现了降低成本的目的,因此猜测可能性不大。
Myc仍然会有天然的干扰,应用略有局限;相较后两者是人工合成专门用于tagged蛋白(有相应的专利,因此目前无论抗体或交联好的beads价格都非常昂贵),因此干扰最小、最常用。如需进一步细致区分,a-Flag效价比a-HA略高,因此更适合IP。当然,公司仍在努力寻找效价更好的a-HA以及IP HA的抗体(Flag系Sigma专利,因此目前只能忍受其过高的定价)。那么,之前颇流行的a-HA鼠源单抗(其clone名称为12CA5)为何被潜在地摒弃了?
原因大概是:该克隆株可以轻易获取,流传甚广,因此可以取腹水自制;效价不高,特异性很差。尤其是特异性的问题,用该抗体去交联的beads做coIP,隐患很大(可以轻易PD非常浓的非特异性蛋白)——因此,购买商品化a-HA beads时,请仔细阅读产品说明(避开12CA5系列)。
实际上,在构建外源过表达体系的时候,通常可以得到一系列表达水平差异的稳定单克隆,可以通过进一步筛选获得外源与内源相加与原内源蛋白水平相当的细胞株(在细胞调控承受的范围以内,内源性蛋白水平会因为相应的外源蛋白表达而适当下调),这样的细胞株可以用于模拟内源性蛋白的相互作用;因为理论上未改变细胞的生理特异,相应的生命过程仍受内源因素调控。
当然构建相应的细胞株需要转染并筛选,又因为要控制表达量水平,筛选工作耗时更长;并且细胞状态会因为传代过多而有些改变。因此,绝对的内源性蛋白的相互作用仍是一个不可或缺的数据,尤其对一个新发现的蛋白复合体。
1. IP外源tagged-A蛋白,洗脱尽量用相应的peptide,一方面提高特异性,另一方面由于洗脱条件温和,重链和轻链脱落也会较少;在IP前,尤其对于新Flag、HA beads可以用washing buffer或者低pH Glycine-HCl漂洗一下beads,把结合不牢的重链和轻链先洗掉。此外,Flag、HA-beads通常是鼠抗,那么IB B蛋白的抗体应该选用兔抗。
即使交错抗体的种属,实际上当重轻链脱落过多时(尤其是重链),在IB的时候它符合《WB实战指南》提到的杂蛋白过浓会非特异结合任意抗体,这在coIP的实验结果分析中要格外小心。有人会说可以用未免疫的IgG做阴性对照,但是偶尔会发生一些未知意外,恰好IgG的重链没有显影(毕竟这不是抗原抗体特异性结合)。
BTW:偶尔这样的结果还能重复出来,但是反过来IP B蛋白,IB A蛋白可能就100%失败。所以,当蛋白分子量接近重、轻链的时候,下结论要格外小心,除严格阴性、阳性对照外,reverse IP的结果也很重要。最好加一个不相干的蛋白的同种属的抗体做阴性对照
抗体剂量上文提过了;其他flag、protein A等等beads一般用10ul足够了,偶尔根据需要微调,诸如过表达纯化蛋白。总之,一般CE的成本较低,若用CE饱和抗体、beads,只要你抗体、beads和操作始终一致,最后IP的A蛋白能保证一致,结果有效。
干粉状的beads用IP buffer充分溶胀后离心去beads的碎片;其他液态的不需要此过程。现在市售的商品化beads如sigma flag可不需要IP buffer漂洗直接IP,未发现对coIP结果的明显干扰。通常,再生的beads由于放置-20度延长保存期需要加入大量甘油,不漂洗直接使用不容易把beads分匀,从而造成不同tube间初始差异,这时候会对IP结果影响很大。
那么,beads翻转越久蛋白会沉淀越多,而这种沉淀无法靠漂洗去除干净,最终进入sample从而可以检测到任意蛋白的结合。因此,限制beads翻转孵育时间非常关键;通常beads孵育1-2hr已经充分结合了。
商品化的beads尤其抗体交联的,因为抗体和专利的原因价格相对昂贵,那么回收beads并再生就显得非常必要。保管得好(防霉变),beads可以重复用20-30次。
再生beads没有什么特别的,常做生化柱层析的,那简直就是家常便饭。IP用beads的再生,也无非就是高盐接酸洗再高盐,最后用IP漂洗buffer复性,加甘油和NaN3扔-20度长期保存。
高盐即3M NaCl;酸洗,就是可用做elution buffer的0.1M pH3.0的Glycine-HCl。IP beads尤其抗体交联的,由于昂贵再加实验和回收过程的损失一般体积不大,所以Biorad公司一种小型塑料的柱层析管用来再生beads最合适(它是一次性的,但仅仅用于再生beads可以无限重复使用);而常规的玻璃生化层析柱即便最小号的也太大,黏壁损失会很大,beads再生几次可能就全损失了。
再生操作就类似做DNA大抽的KIT,加液体到层析柱、控制流速让其一滴滴流下;再生质量可以通过改变酸、盐的体积控制,希望干净点多加一些、多洗几遍。
因此,对于静置很久的旧beads通常在回收前一天高速翻转过夜,这时候再把beads转移到层析柱中就会相对疏松,酸、盐洗涤就会比较流畅。同样的原因,再生过程中通常就是流速越来越慢,再生次数多的旧beads其内含的灰尘、颗粒也较多,可能最后液体就不滴了;这时候只能用枪反复吹吸、松松beads。当然,这些问题都只在beads体积较大的时候时明显,如果本来就不足1ml,以上就全是废话。