分子进化树构建及数据分析的简介
互联网
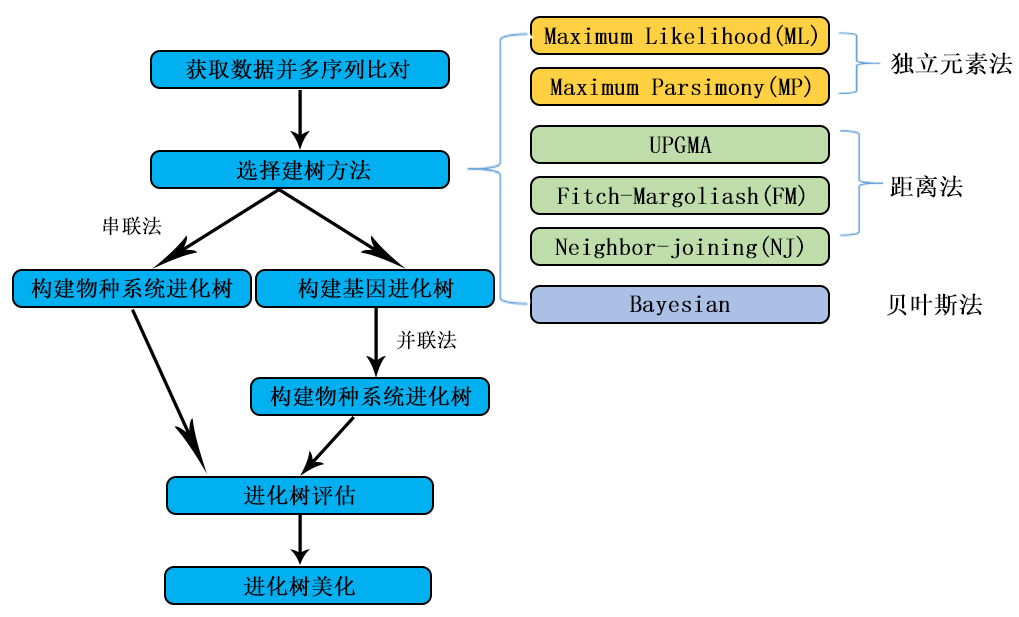
一、方法的选择
首先是方法的选择。基于距离的方法有UPGMA、ME(Minimum Evolution,最小进化法)和NJ(Neighbor-Joining,邻接法)等。其他的几种方法包括MP(Maximum parsimony,最大简约法)、ML(Maximum likelihood,最大似然法)以及贝叶斯(Bayesian)推断等方法。其中UPGMA法已经较少使用。
一般来讲,如果模型合适,ML的效果较好。对近缘序列,有人喜欢MP,因为用的假设最少。MP一般不用在远缘序列上,这时一般用NJ或ML.对相似度很低的序列,NJ往往出现Long-branch attraction(LBA,长枝吸引现象),有时严重干扰进化树的构建。贝叶斯的方法则太慢。对于各种方法构建分子进化树的准确性,一篇综述(Hall BG. Mol Biol Evol 2005,22(3):792-802)认为贝叶斯的方法最好,其次是ML,然后是MP.其实如果序列的相似性较高,各种方法都会得到不错的结果,模型间的差别也不大。
对于NJ和ML,是需要选择模型的。对于各种模型之间的理论上的区别,这里不作深入的探讨,可以参看Nei的书。对于蛋白质序列以及DNA序列,两者模型的选择是不同的。以作者的经验来说,对于蛋白质的序列,一般选择Poisson Correction(泊松修正)这一模型。而对于核酸序列,一般选择Kimura 2-parameter(Kimura-2参数)模型。如果对各种模型的理解并不深入,作者并不推荐初学者使用其他复杂的模型。
Bootstrap几乎是一个必须的选项。一般Bootstrap的值>70,则认为构建的进化树较为可靠。如果Bootstrap的值太低,则有可能进化树的拓扑结构有错误,进化树是不可靠的。
对于进化树的构建,如果对理论的了解并不深入,作者推荐使用缺省的参数。需要选择模型的时候(例如用NJ或者ML建树),对于蛋白序列使用Poisson Correction模型,对于核酸序列使用Kimura-2参数模型。另外需要做Bootstrap检验,当Bootstrap值过低时,所构建的进化树其拓扑结构可能存在问题。并且,一般推荐用两种不同的方法构建进化树,如果所得到的进化树类似,则结果较为可靠。
二、软件的选择
| 软件 | 网址 | 说明 |
| ClustalX | http://bips.u-strasbg.fr/fr/Documentation/ClustalX/ | 图形化的多序列比对工具 |
| ClustalW | http://www.cf.ac.uk/biosi/research/biosoft/Downloads/clustalw.html | 命令行格式的多序列比对工具 |
| GeneDoc | http://www.psc.edu/biomed/genedoc/ | 多序列比对结果的美化工具 |
| BioEdit | http://www.mbio.ncsu.edu/BioEdit/bioedit.html | 序列分析的综合工具 |
| MEGA | http://www.megasoftware.net/ | 图形化、集成的进化分析工具,不包括ML |
| PAUP | http://paup.csit.fsu.edu/ | 商业软件,集成的进化分析工具 |
| PHYLIP | http://evolution.genetics.washington.edu/phylip.html | 免费的、集成的进化分析工具 |
| PHYML | http://atgc.lirmm.fr/phyml/ | 最快的ML建树工具 |
| PAML | http://abacus.gene.ucl.ac.uk/software/paml.html | ML建树工具 |
| Tree-puzzle | http://www.tree-puzzle.de/ | 较快的ML建树工具 |
| MrBayes | http://mrbayes.csit.fsu.edu/ | 基于贝叶斯方法的建树工具 |
| MAC5 | http://www.agapow.net/software/mac5/ | 基于贝叶斯方法的建树工具 |
| TreeView | http://taxonomy.zoology.gla.ac.uk/rod/treeview.html | 进化树显示工具 |
表1中列出了一些与构建分子进化树相关的软件构建NJ树,可以用PHYLIP(写得有点问题,例如比较慢,并且Bootstrap检验不方便)或者MEGA.MEGA是Nei开发的方法并设计的图形化的软件,使用非常方便。作者推荐MEGA软件为初学者的首选。虽然多雪列比对工具ClustalW/X自带了一个NJ的建树程序,但是该程序只有p-distance模型,而且构建的树不够准确,一般不用来构建进化树。
构建MP树,最好的工具是PAUP,但该程序属于商业软件,并不对学术免费。因此,作者并不建议使用PAUP。而MEGA和PHYLIP也可以用来构建进化树。这里,作者推荐使用MEGA来构建MP树。理由是,MEGA是图形化的软件,使用方便,而PHYLIP则是命令行格式的软件,使用较为繁琐。对于近缘序列的进化树构建,MP方法几乎是最好的。
构建ML树可以使用PHYML,速度最快。或者使用Tree-puzzle,速度也较快,并且该程序做蛋白质序列的进化树效果比较好。而PAML则并不适合构建进化树。ML的模型选择是看构出的树的likelihood值,从参数少,简单的模型试起,到likelihood值最大为止。ML也可以使用PAUP或者PHYLIP来构建。这里作者推荐的工具是BioEdit。BioEdit集成了一些PHYLIP的程序,用来构建进化树。Tree-puzzle是另外一个不错的选择,不过该程序是命令行格式的,需要学习DOS命令。PHYML的不足之处是没有win32的版本,只有适用于64位的版本,因此不推荐使用。值得注意的是,构建ML树,不需要事先的多序列比对,而直接使用FASTA格式的序列即可。
贝叶斯的算法以MrBayes为代表,不过速度较慢。一般的进化树分析中较少应用。由于该方法需要很多背景的知识,这里不作介绍。
需要注意的几个问题是,其一,如果对核酸序列进行分析,并且是CDS编码区的核酸序列,一般需要将核酸序列分别先翻译成氨基酸序列,进行比对,然后再对应到核酸序列上。这一流程可以通过MEGA 3.0以后的版本实现。MEGA3现在允许两条核苷酸,先翻成蛋白序列比对之后再倒回去,做后续计算。其二,无论是核酸序列还是蛋白序列,一般应当先做成FASTA格式。FASTA格式的序列,第一行由符号“>”开头,后面跟着序列的名称,可以自定义,例如user1,protein1等等。将所有的FASTA格式的序列存放在同一个文件中。文件的编辑可用Windows自带的记事本工具,或者EditPlus(google搜索可得)来操作。文件格式如图1所示:

图1 FASTA格式的序列
另外,构建NJ或者MP树需要先将序列做多序列比对的处理。作者推荐使用ClustalX进行多序列比对的分析。多序列比对的结果有时需要后续处理并应用于文章中,这里作者推荐使用GeneDoc工具。而构建ML树则不需要预先的多序列比对。
因此,作者推荐的软件组合为:MEGA 3.1 + ClustalX + GeneDoc + BioEdit。
三、数据分析及结果推断
一般碰到的几类问题是:
(1)推断基因/蛋白的功能;
(2)基因/蛋白家族分类;
(3)计算基因分化的年代。
关于这方面的文献非常多,这里作者仅做简要的介绍。
推断基因/蛋白的功能,一般先用BLAST工具搜索同一物种中与不同物种的同源序列,这包括直向同源物(ortholog)和旁系同源物(paralog)。如何界定这两种同源物,网上有很多详细的介绍,这里不作讨论。然后得到这些同源物的序列,做成FASTA格式的文件。一般通过NJ构建进化树,并且进行Bootstrap分析所得到的结果已足够。如果序列近缘,可以再使用MP构建进化树,进行比较。如果序列较远源,则可以做ML树比较。使用两种方法得到的树,如果差别不大,并且Bootstrap总体较高,则得到的进化树较为可靠。
基因/蛋白家族分类。这方面可以细分为两个问题。一是对一个大的家族进行分类,另一个就是将特定的一个或多个基因/蛋白定位到已知的大的家族上,看看属于哪个亚家族。例如,对驱动蛋白(kinesin)超家族进行分类,属于第一个问题。而假如得到一个新的驱动蛋白的序列,想分析该序列究竟属于驱动蛋白超家族的14个亚家族中的哪一个,则属于后一个问题。这里,一般不推荐使用MP的方法。大多数的基因/蛋白家族起源较早,序列分化程度较大,相互之间较为远源。这里一般使用NJ、ME或者ML的方法。
计算基因分化的年代。这个一般需要知道物种的核苷酸替代率。常见物种的核苷酸替代率需要查找相关的文献。这里不作过多的介绍。一般对于这样的问题,序列多数是近缘的,选择NJ或者MP即可。
如果使用MEGA进行分析,选项中有一项是“Gaps/Missing Data”,一般选择“Pairwise Deletion”。其他多数的选项保持缺省的参数。
四、总结
在实用中,只要方法、模型合理,建出的树都有意义,可以任意选择自己认为好一个。最重要的问题是:你需要解决什么样 的问题?如果分析的结果能够解决你现有的问题,那么,这样的分析足够了。因此,在做进化分析前,可能需要很好的考虑一下自己的问题所在,这样所作的分析才有针对性。