DNA甲基化研究方法的回顾与评价(图)
互联网
摘要: DNA 甲基化是表观遗传学(Epigenetics)的重要组成部分,在维持正常细胞功能、遗传印记、胚胎发育以及人类肿瘤发生中起着重要作用,是目前新的研究热点之一。随着对甲基化研究的不断深入,各种各样甲基化检测方法被开发出来以满足不同类型研究的要求。这些方法概括起来可分为三类:整体水平的甲基化检测、基因特异位点甲基化的检测和新甲基化位点的寻找。本篇将主要介绍目前存在的大部分DNA 甲基化研究方法,并对其相关特性进行了简要分析与总结。
关键词 :表观遗传学;DNA 甲基化;甲基化研究方法
早在1942年,C.H.Waddington首次提出表观遗传学(epigenetics)的概念,并指出表观遗传与遗传是相对的,它主要研究基因型和表型的关系。几十年后,霍利迪(R.Holiday)针对表观遗传学提出了更新的系统性论断,也就是人们现在比较统一的认识[1],即在不改变基因组序列的前提下,通过DNA 和组蛋白的修饰来调控基因表达,这种修饰以DNA 甲基化最为常见。继人类基因组计划结束后,2003年人类表观基因组协会(Human Epigenome Consortium,HEC)宣布开始投资和实施人类表观基因组计划(HEP)。
其主要任务是绘制出人类中甲基化可变位点图谱,即不同组织与疾病状态下,5-甲基胞嘧啶出现及其分布频率的图谱,以指导和系统地研究DNA 甲基化在人类表观遗传、胚胎发育、基因印记、等位基因失活及肿瘤发生中的重要作用[2]。DNA 甲基化的研究,逐渐成为新的研究热点。随着对甲基化研究的不断深入,各种各样甲基化检测方法被开发出来以满足不同类型研究的要求。让我们一一介绍现有的大部分DNA 甲基化研究方法,并对其相关特性进行简要分析与总结。
1导言
1.1 DNA 甲基化及CpG岛
DNA 甲基化是最早发现的基因表观修饰方式之一,可能存在于所有高等生物中。DNA 甲基化能关闭某些基因的活性,去甲基化则诱导了基因的重新活化和表达。甲基化的主要形式有5-甲基胞嘧啶,N6-甲基腺嘌呤和7-甲基鸟嘌呤。原核生物中CCA/TGG和GATC常被甲基化,而真核生物中甲基化仅发生于胞嘧啶。DNA 的甲基化是在DNA 甲基化转移酶(DNMTs)的作用下使CpG二核苷酸5'端的胞嘧啶转变为5'甲基胞嘧啶。这种DNA 修饰方式并没有改变基因序列,但是它调控了基因的表达[3]。脊椎动物基因的甲基化状态有三种:持续的低甲基化状态,如管家基因;去甲基化状态,如发育阶段中的一些基因;高度甲基化状态,如女性的一条失活的X染色体[4]。
哺乳动物中,CpG序列在基因组中出现的频率仅有1%,远低于基因组中的其它双核苷酸序列。但在基因组的某些区域中,CpG序列密度很高,可以达均值的5倍以上,成为鸟嘌呤和胞嘧啶的富集区,形成所谓的CpG岛[5]。通常,CpG岛大约含有500多个碱基。
在哺乳动物基因组中约有4万个CpG岛,而且只有CpG岛的胞嘧啶能够被甲基化[6],CpG岛通常位于基因的启动子区或是第一个外显子区[7]。健康人中,CpG岛中的CpG位点通常是处于非甲基化状态,而在CpG岛外的CpG位点则通常是甲基化的。这种甲基化的形式在细胞分裂的过程中能够稳定的保留[8]。当肿瘤发生时,抑癌基因CpG岛以外的CpG序列非甲基化程度增加,而CpG岛中的CpG则呈高度甲基化状态,以致于染色体螺旋程度增加及抑癌基因表达的丢失[9]。
1.2 DNA 甲基化的生物学作用
1.2.1 DNA 甲基化与遗传印记、胚胎发育
DNA 甲基化在维持正常细胞功能、遗传印记、胚胎发育过程中起着极其重要的作用。研究表明胚胎的正常发育得益于DNA 适当的甲基化。例如:缺少任何一种甲基转移酶对小鼠胚胎的发育都是致死性的(Li等1992年和Okano等1999年)[3]。此外,等位基因的抑制(allelic repression)被印记控制区(imprinting control regions,ICRs)所调控,该区域在双亲中的一个等位基因是甲基化的[4]。印记基因的异常表达可以引发伴有突变和表型缺陷的多种人类疾病。如:脐疝-巨舌-巨大发育综合征(Beckwith-Wiedemann Syndrome,BWS)和Prader-Willi/Angelman综合征等[10]。
1.2.2 DNA 甲基化与肿瘤
甲基化状态的改变是引起肿瘤的一个重要因素,这种变化包括基因组整体甲基化水平降低和CpG岛局部甲基化水平的异常升高,从而导致的不稳定(如染色体的不稳定、可移动遗传因子的激活、原癌基因的表达)[4]和抑癌基因的不表达。如果抑癌基因中有活性的等位基因失活,则发生癌症的机率提高,例如:胰岛素样生长因子-2(IGF-2)基因印记丢失导致多种肿瘤,如Wilm‘s瘤[11]。
目前肿瘤甲基化的研究主要集中在抑癌基因。这是因为人们发现肿瘤的发生可能与抑癌基因启动子区的CpG岛甲基化造成抑癌基因关闭有关[12]。由于CpG岛的局部高度甲基化早于细胞的恶性增生,因此甲基化的诊断可以用于肿瘤发生的早期预测[13],而且全的低甲基化也随着肿瘤发生而出现,并且其随着肿瘤恶性度的增加而显著[14] ,因此甲基化的检测可用于肿瘤的分级。
Shinichi Toyooka描述了肿瘤发生与异常甲基化的关系:被SV40 (Simian Virus 40)感染的人间皮细胞,其端粒酶活性上调,Notch-1基因表达增加,肿瘤相关基因(包括抑癌基因RASSF1A)的启动子区发生异常甲基化[15]。Cui等发现部分结肠癌患者的正常肠粘液腺细胞的IGF-2基因印记丢失[16]。Uhlmann等发现不同病理类型及不同恶性程度的神经胶质瘤细胞的7种肿瘤标志基因存在着不同程度的甲基化 [17]。因此,甲基化的研究,为肿瘤的早期预测、分类、分级及预后评估提供了新的依据 。
1.3 DNA 甲基化的研究方法
近15年来,人们越来越认识到DNA 甲基化研究的重要性,开发出一系列检测DNA 的方法。根据研究目的这些方法分为:整体水平的甲基化检测,特异位点甲基化的检测和新甲基化位点的寻找。根据研究所用处理方法不同可以分为:基于PCR的甲基化分析方法;基于限制性内切酶的甲基化分析方法;基于重亚硫酸盐的甲基化分析方法和柱层法等。Christina Dahl和Per Guldberg[3]归纳总结了主要的甲基化分析方法及相关特性,在此基础上我们略加以补充(见表1)。
2 甲基化研究方法学回顾
2.1 整体水平甲基化分析
2.1.1 高效液相色谱 柱(HPLC)及相关方法
HPLC是一种比较传统的方法,能够定量测定整体水平DNA 甲基化水平。它由Kuo等1980年[18]首次报道。过程是将DNA 样品先经盐酸或氢氟酸水解成碱基,水解产物通过色谱 柱,结果与标准品比较,用紫外光测定吸收峰值及其量,计算5m C/(5m C+5℃)的积分面积就得到基因组整体的甲基化水平。这是一种检测DNA 甲基化的标准方法。但它需要较精密的仪器。Fraga等2002年[19]运用高效毛细管电泳法(HPCE)处理DNA 水解产物,以确定5mC的水平。与HPLC相比,HPCE更加简便、快速、经济。HPLC及HPCE测定整体DNA 甲基化水平的敏感性均较高。Oefner等1992年[20]提出变性高效液相色谱 法(DHPLC)用于分析单核苷酸和DNA 分子。邓大君等2001[21]将其改进与PCR联用建立了一种检测甲基化程度的DHPLC分析方法。将重亚硫酸盐处理后的产物进行差异性扩增,由于原甲基化的在重亚硫酸盐处理时仍被保留为胞嘧啶,因此原甲基化的在PCR扩增时,其变性温度也相应上升,使PCR产物在色谱 柱中保留的时间明显延长,这样就可以测定出PCR产物中甲基化的情况。
这种方法的最明显优点是:可用于高通量混合样本检测,能够明确显示目的片段中所有CpG位点甲基化的情况,但不能对甲基化的CpG位点进行定位。
2.1.2 SssI 甲基转移酶法[22]
SssI甲基转移酶能够催化DNA 的CpG位点发生甲基化。3 H-S-腺苷甲硫氨酸(3 H-SAM)在SssI甲基转移酶催化作用使基因组DNA 的CpG位点发生甲基化。通过测定剩余的放射性标记的SAM即可得到原整体甲基化水平,即测到的放射性强度与所测DNA 甲基化水平成反比。这种方法的缺点是所使用的SssI甲基转移酶不稳定,致结果不够精确。
2.1.3 免疫化学法[23]
这种方法是基于单抗体能够与5m C发生特异性反应。应用荧光素标记抗体使之与预先已固定在DEAE膜上的样品DNA 特异性结合,对DEAE膜上的荧光素进行扫描得到5m C的水平,其荧光素强度与5m C水平成正比。Oakeley等1997年[23]报道了这种方法。这种方法需要精密的仪器。
2.1.4 氯乙醛法
Oakeley等1999年[24]首先描述了这种使用氯乙醛和荧光标记的方法。首先,将DNA 经重亚硫酸盐处理使未甲基化的胞嘧啶全部转变为尿嘧啶,而甲基化的胞嘧啶保持不变(Frommer等1992年)[25],然后经过银或色谱 柱去除DNA 链上的嘌呤,再将样品与氯乙醛共同孵育,这样5m C就转变为带有强荧光的乙烯胞嘧啶,荧光的强度与原5m C的水平成正比。这种方法可以直接测定整体5m C水平。其优点是所用试剂价格低廉且稳定性好,避免了放射性污染,但缺点是费时费力,而且氯乙醛是一种有毒的物质。
2.2 特异性位点的DNA 甲基化的检测
2.2.1 甲基化敏感性限制性内切酶(methylation-sensitive restriction Endonuclease,MS-RE)-PCR/Southern法
这种方法利用甲基化敏感性限制性内切酶对甲基化区的不切割的特性,将DNA 消化为不同大小的片段后再进行分析。常使用的甲基化敏感的限制性内切酶有HpaⅡ-MspⅠ(识别序列CCGG)和SmaⅠ-Xmal(CCCGGG)等。由于后者识别的碱基数相对较多,其碱基序列在体内出现的概率相对较低,所以以前者即HpaⅡ-MspⅠ更常用。其中HpaⅡ和MspⅠ均能识别CCGG序列,然而当序列中的胞嘧啶发生甲基化时,HpaⅡ不切割,利用HpaⅡ-MspⅠ的这种属性处理DNA ,随后进行Southern或PCR扩增分离产物,明确甲基化状态[12][26]。
这是一种经典的甲基化研究方法,其优点是:相对简单,成本低廉,甲基化位点明确,实验结果易解释;缺点是:1.由于CG不仅仅限于CCGG序列中,因此非该序列中的CG将被忽略;2.只有检测与转录相关的关键性位点的甲基化状态时,该检测方法的结果才有意义;3.相对而言,Southern方法较复杂,且需要样本的量大;4.存在着酶不完全消化引起的假阳性的问题;5.不适用于混合样本。
2.2.2 直接测序 法
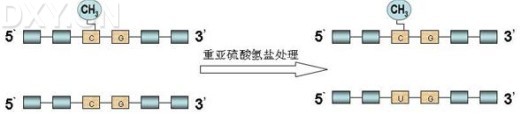
直接测序 是由Frommer等[25]提出的研究DNA 甲基化方法。过程是:重亚硫酸盐使DNA 中未发生甲基化的胞嘧啶脱氨基转变成尿嘧啶,而甲基化的胞嘧啶保持不变(见图1),行PCR扩增所需片段,则尿嘧啶全部转化成胸腺嘧啶,最后,对PCR产物进行测序 并且与未经处理的序列比较,判断是否CpG位点发生甲基化。此方法是一种可靠性及精确度很高的方法,能明确目的片段中每一个CpG位点的甲基化状态,但需要大量的测序 ,过程较为繁琐、昂贵[27]。

图1:重亚硫酸盐处理过程示意图。DNA 经重亚硫酸盐处理后,甲基化的胞嘧啶不变,未甲基化的胞嘧啶转变为尿嘧啶
2.2.3 甲基化特异性的PCR(methylation-specific PCR,MS-PCR)
Herman等1996年[28]在使用重亚硫酸盐处理的基础上新建的一种方法。它将DNA 先用重亚硫酸盐处理,这样未甲基化的胞嘧啶转变为尿嘧啶,而甲基化的不变,随后行引物特异性的PCR。MS-PCR中设计两对引物,并要求:1.引物末端均设计至检测位点结束;2.两对引物分别只能与重亚硫酸盐处理后的序列互补配对,即一对结合处理后的甲基化DNA 链,另一对结合处理后的非甲基化DNA 链。检测MSP扩增产物,如果用针对处理后甲基化DNA 链的引物能扩增出片段,则说明该被检测的位点存在甲基化;若用针对处理后的非甲基化DNA 链的引物扩增出片段,则说明被检测的位点不存在甲基化(见图2)[26][27]。

图2:甲基特异性的PCR扩增(MS-PCR)示意图。DNA 经重亚硫酸盐处理后,以处理后的产物作为模板,加入甲基化特异性的引物(primerⅠ)或非甲基化的引物(primerⅡ),进行特异性的扩增(如图所示),只有结合完全的甲基化或非甲基化特异性引物的片段才能扩增出产物。
这种方法的优点是:1.避免了使用限制性内切酶及其后续相关问题;2.敏感性高;可用于石蜡包埋样本[12];缺点是:1.要预先知道待测片段DNA 的序列;2.引物设计至关重要;3.若待测DNA 中5-甲基胞嘧啶分布极不均衡,则检测时较为复杂;4.这种方法只能作定性研究,即只能明确是否存在甲基化;若要求定量,则需用其他的方法进行进一步检测;5.存在重亚硫酸盐处理不完全导致的假阳性。
2.2.4 甲基化敏感性单核苷酸引物延伸(methylation-sensitive single nucleotide primer extension,Ms-SnuPE)
Gonzalgo and Jones 1997年提出了结合重亚硫酸盐处理和单核苷酸引物延伸(Kuppuswamy等1991年提出[29])的Ms-SnuPE方法[30],用于定量检测已知序列中特异位点的甲基化水平。过程是:先将研究序列用重亚硫酸盐处理,未甲基化的胞嘧啶全部转化为尿嘧啶,而甲基化的胞嘧啶不变。进行PCR扩增,然后取等量扩增产物置于2管中,分别作为Ms-SnuPE单核苷酸引物延伸的模板。设计用于Ms-SnuPE延伸的引物的3’端紧邻待测碱基。同时于2个反应体系中加入等量的Taq酶、引物、同位素标记的dCTP或dTTP。这样,如果待测位点被甲基化,则同位素标记的dCTP会在反应延伸时连于引物末端;若是未被甲基化,则标记的dTTP参与反应。末端延伸产物经电泳分离和放射活性测定后可得出C/T值,即为甲基化与非甲基化的比值,从而分析得到待测片段中CpG位点甲基化情况[5][29](见图3)。同理也可以用dGTP或dATP。而且,若需研究一条链上不同位点CpG甲基化情况,可通过设计不同的引物在同一反应中完成[12]。
图3: 甲基化敏感性单核苷酸引物延伸(Ms-SnuPE)示意图。DNA 经重亚硫酸盐处理后,以PCR扩增后得到产物作为Ms-SnuPE延伸的模板,于反应体系中入设计好的引物和同位素标记的dCTP或dTTP进行甲基化特异的单核苷酸引物延伸,随后,电泳分离、测定同位素放射活性,确定甲基化水平。
这种方法的优点:1.可以了解特异位点甲基化情况且不受内切酶的限制;2.通过设计的不同引物在同一延伸反应情况可以了解不同位点CpG甲基化的状况;3.可以检测出样本序列中分布不均匀的甲基化位点;4.是一种能够用于定量检测甲基化水平的方法;5.仅需少量的DNA 样本,可以用于石蜡包埋样本的测定。缺点是:1.实验步骤略复杂,若要检测多个位点时则需设计多个引物[5];2.存在放射性污染及重亚硫酸盐处理不完全的问题。
2.2.5 结合重亚硫酸盐的限制性内切酶法(combined bisulfite restriction analysis,COBRA)
Xiong and Peter报道了COBRA[31]甲基化检测法。这种方法对标本DNA 行重亚硫酸盐处理及PCR扩增,处理后原甲基化的胞嘧啶被保留,而非甲基化的胞嘧啶变为胸腺嘧啶。随后用限制性内切酶对转化后PCR产物切割的特性以识别原标本DNA 的甲基化状况。(见图4)。

图4: 结合重亚硫酸盐的限制性内切酶法(COBRA)示意图。重亚硫酸盐处理DNA 后行PCR扩增,用限制性内切酶(BstUI)识别转化后序列中的酶切位点,消化产物电泳分离,与完全非甲基化阴性对照组比较,得出序列中特异位点甲基化水平(图4参考引文[31]并略加修改)。
这种方法的优点有:1.方法相对简单,不需预先知道CpG位点及样本序列;2.可以进行甲基化水平的定量研究;3.需要样本量少,可用于石蜡包埋样本的分析[32]。缺点是:1.只能获得特殊酶切位点甲基化情况,因此检测阴性不能排除样品DNA 中存在甲基化的可能[5];2.由于酶和PCR的使用,只能分析一种特定序列[5]。
2.2.6 甲基化敏感性单链构象分析(methylation-specific single-strand conformation analysis,MS-SSCA)
甲基化敏感性单链构象分析(MS-SSCA)又称重亚硫酸盐甲基化-PCR-SSCP(Single-Strand Conformation Polymorphism,SSCP)(BiPS),由Maekawa等1999年[32]报道。方法是:先用重亚硫酸盐处理待测片段,针对非CG二核苷酸区设计引物进行PCR扩增,扩增产物变性后作非变性的聚丙酰胺凝胶电泳,由于DNA 电泳时的移动性取决于其二级结构即DNA 的空间构象,而后者又由DNA 碱基的序列决定。因此,经处理后变性的单链DNA 将停留在聚丙酰胺膜的不同位置上,这样甲基化与非甲基化的就被分离开,随后行单链构象多态性分析加以明确(见图5):

图5:甲基化敏感性单链构象分析(MS-SSCA)示意图。重亚硫酸盐处理DNA 后,设计引物(非CG二核苷酸区)对处理后的DNA 进行扩增,产物解链后行聚丙酰胺凝胶电泳,由于甲基化和非甲基化单链DNA 形成不同的空间构象,它们在电泳中移动速率不同,故出现在不同位置,从而判定待测片段中甲基化情况。
这种方法的优点是:1.能够方便的应用于任何序列的甲基化状态分析;2.能够对甲基化的等位基因进行半定量;3.可以提示甲基化状态分布的不均匀性;缺点是:1.只有甲基化水平较高的单链才能明显的区分开,而较低水平的则不易分开,有时会因甲基化的CpG位点随机和不均匀分布导致电泳条带出现拥挤、拖尾的现象,故敏感性及准确性略低;2.检测片段不宜过长。
2.2.7 甲基化敏感性变性梯度凝胶电泳(methylation-specific denaturing gradient gel electrophoresis,MS-DGGE)
变性梯度凝胶电泳(DGGE)是一种能够将具有单碱基差别的DNA 分离的方法。其原理是:当双链DNA 在变性梯度凝胶中进行到与DNA 变性温度对应一致的位置时,DNA 部分解链(解链区域的长度大小不等),与每一个解链区域相对应的温度称为解链温度(T)。Tm 主要由核苷酸的序列决定,这是因为DNA 链上相邻碱基间的相互作用对稳定DNA 双螺旋起重要作用。因此,很小的变化(如单碱基变化)也会引起DNA 片段Tm 值的改变。DGGE系统中,DNA 片段在变性梯度聚丙烯酰胺凝胶电泳时,由于凝胶中变性剂浓度自上而下呈梯度递增,因此,当DNA 片段到达与该区域的T值相当的某一浓度位置时,DNA 解链变为分枝状,其移动减慢,停留在凝胶的的某一位置,这样不同的DNA 片段就被分离。mAggerholm等1999年[35]将其用于甲基化的检测,先用重亚硫酸盐处理DNA 使为甲基化的胞嘧啶转变为尿嘧啶引起点突变,这样再结合使用DGGE,经电泳分离、分析该片段的甲基化状况。
这种方法的优点是:DGGE可以用来检测出除最高温度解链区域以外的所有发生甲基化的DNA 片段,需样品量少,能较直观的显示出甲基化情况[36]。缺点是:解链温度和DGGE的变性浓度梯度需要摸索。
2.2.8 甲基化敏感性解链曲线分析(methylation-specific melting curve analysis,MS-MCA)
Worm等[37]2001年报道的MS-MCA是将DNA 经重亚硫酸盐处理与Lightcycle联用检测DNA 序列甲基化的方法。荧光素标记双链DNA 。这种方法根据检测到的荧光度对应的解链温度,判断分析研究序列中甲基化的情况。在Lightcycle过程中,随着温度升高,逐渐达到DNA 双链各解链区域的解链温度Tm,DNA 呈区域性逐渐解链,一般说来,序列中CG含量越高,对应的解链温度越高。由于非甲基化的胞嘧啶经重亚硫酸盐处理后变为尿嘧啶、PCR后变为胸腺嘧啶,故其所在序列中的CG含量降低,热稳定性降低,解链温度降低。而甲基化
的由于其CG含量高,故其解链温度高。所作结果与标准曲线对照,根据这种特性就可以明确研究序列中CpG的分布区及甲基化程度。(见图6、图7)。

图6:甲基化敏感性解链曲线分析(MS-MCA)示意图。完全非甲基化时,解链温度低(如A);完全甲基化时,解链温度高(如B);当等位基因的甲基化的发生集中于一条链或等位基因甲基化嵌合分布时,解链温度改变(如C、D)(图6参考引文 [37])。

图7:甲基化程度与解链温度关系示意图。p15Ink4b基因的荧光解链曲线,重亚硫酸盐处理HL-60(曲线a,完全未甲基化的)和MOLT-4(曲线b,完全甲基化的)这两个细胞系及取自慢性髓性白血病病人骨髓细胞中p15Ink4b的基因(其曲线为c和d,示部分甲基化的)。曲线a、b、c、d的解链峰值分别为81.3℃、88.9℃、84.4℃和86.2℃(图7参考引文 [37])。
这是一种能对甲基化分布不均匀的DNA 样本进行半定量分析的方法。缺点是:1.它不能够精确检测甲基化的具体位点;2.研究序列的长度不宜过长;3.该法对低水平的DNA 甲基化敏感性低[37]。
2.2.9 荧光法(Methylight)
Eads等[38]2000年报道的荧光法利用实时PCR(Real-time PCR)测定特定位点甲基化的情况。其过程如下:先用重亚硫酸盐处理待测DNA 片段。设计一个能与待测位点区互补的探针,探针的5'端连接报告荧光,3'端连接淬灭荧光,随后行实时定量PCR。如果探针能够与DNA 杂交,则在PCR用引物延伸时,TaqDNA 聚合酶5'到3'端的外切酶活性会将探针序列上5'端的报告荧光切下,淬灭荧光不再能对报告荧光进行抑制,这样报告荧光发光,测定每个循环报告荧光的强度即可得到该位点的甲基化情况及水平;同理,若标记的探针未能与DNA 杂交,则引物延伸不能跳过未甲基化位点,报告荧光不被切下,不发光。同样方法,也可对引物进行荧光标记,并通过不同标记的组合,检测多个位点的甲基化水平[39]。
高敏感、快速是本方法最显著的特点,它可以在非甲基化等位基因超出10000倍的情况下精确的检测到甲基化的等位基因并定量,而且可以做多样本、多基因位点的快速分析。此外其具备可重复、所需样本量少、不需要[12]电泳分离的特点。它可以为临床标本的分子生物学研究提供可靠的技术支持。缺点是费用高,测定每个位点都要用两端标有荧光素的探针和一对引物,且受较多因素影响。
2.2.10 DNA 微阵列法
Yan等2001年[40]将以分子杂交为基础的微阵列技术应用于DNA 甲基化检测中,这种方法是基于杂交的寡核苷酸微阵列,是一种在基因组中寻找新位点的方法。包括用于整个范围内扫描的差异甲基化(DMH)杂交(Huang等1999年[41])和用于检测某个位点的甲基化特异性微阵列MSO(Gitan等2002年[42])。前者类似于mRNA 表达谱或cDNA 微阵列,是CpG岛微阵列,后者类似于寡核苷酸微阵列,是针对CpG二核苷酸位点的甲基化特异性寡核苷酸微阵列[26]。
MSO要求预先设计一对含有2个不相邻的GC(或AC)的探针,用于识别甲基化和非甲基化的序列,其中含GC的探针(5[42]'---GC---GC---3')识别甲基化序列,含AC的探针(5'---AC---AC---3')识别非甲基化序列,探针的5'端通过Linker固定于玻璃板上。首先对待研究片段用重亚硫酸盐处理,将非甲基化的胞嘧啶变为尿嘧啶,甲基化的不变,再行PCR扩增,产物的3'端用荧光素标记,移至连有探针的玻璃板上进行杂交,通过检测杂交后产生的荧光强度判断待测序 列中甲基化的水平。此方法一定要设立对照。该法可用于多样本、多位点甲基化的检测,样本需要量少,适于临床样本,但存在假阳性问题。
2.2.11 甲基化敏感性斑点分析(methylation sensitive dot blotassay,MS-DBA)
G Clément等2005年[43]报道了一种新的方法,能够定量或半定量分析样本中的甲基化水平。这个方法的过程是:先用重亚硫酸盐处理待测DNA 片段,随后以非CG区的引物行PCR扩增,将扩增产物变性后转移到尼龙膜上,用3'端DIG标记的含有2个CG(或TG)的双核苷酸探针与DNA 杂交,随后用带有荧光标记的抗DIG抗体与之反应。与双CG的探针获得杂交的标本含有甲基化,而与TG探针杂交的标本未被甲基化。通过比较斑点上荧光的强度测定甲基化水平。
这种方法的优点是快速、简便、易于掌握,可一次检测多种样本,包括石蜡包埋样本。缺点是:1.检测序 列不能过长;2.若探针错误杂交或重亚硫酸盐处理不完全,则可能出现假阳性或假阴性结果。
2.2.12 甲基化特异性多连接依赖性探针扩(Methylation-Specific multiplex ligation-dependent probe amplification,MS-mlPA)
Anders O.H.等2005年改进mlPA[44]为MS-mlPA[45]用于检测特异位点的甲基化。MS-mlPA中,针对甲基化的和非甲基化的位点分别设计两个探针,每个探针都包括两个寡核苷酸部分,其中短的部分由合成产生,长的部分来源于phageM13的衍生物,后者有一个非杂交填充片段,不同探针其长度不同。探针的两个寡核苷酸杂交序列均与目标序列互补,其末端都连有相同的PCR引物。且要求[45]设计的MS-mlPA的探针要有甲基化敏感的限制性内切酶HhaⅠ(GCGC)或HpaⅡ(CCGG)的识别位点,这样,经探针和目标序列杂交后,降低反应体系温度,并向其中加入连接酶HhaⅠ,若原样本DNA 中含有HhaⅠ识别的非甲基化的位点,则非甲基化探针的两个寡核苷酸部分不能连接,而甲基化的探针顺利连接不被切割,在随后的PCR反应中只有两个寡核苷酸部分连接后的探针才能被扩增。最后分析扩增片段,确定待研究位点的甲基化情况(具体过程见图8)。

图8:甲基化特异性多连接依赖性探针扩增(MS-mlPA)示意图。用设计好的甲基化与非甲基化的探针(探针各寡核苷酸部分含有杂交序列,不同探针其填充片段的长短不同)分别与待测DNA 杂交,结合上的寡核苷酸探针经连接酶连接,若DNA 片段中存在甲基化位点,则带有(HhaⅠ)识别位点的探针不能被切割,同理,若为非甲基化,则DNA 片段被HhaⅠ切开,且探针不能被连接,这样,在随后的PCR扩增过程中,只有连接的探针能被扩增,最后对扩增的片段进行分析,得出原DNA 中特异位点甲基化的情况(图8参考引文[45])。
这种方法的优点是:1.需要样本的数量少,由于探针具有非常短的识别序列,因此MPLA可以用于局部降解的DNA ,如从石蜡包埋的DNA 样本;2.可用于分析大量混合样本。缺点是探针连接位点受限制性内切酶位点识别的限制,且要考虑到所用酶的反应适宜温度。
2.3甲基化新位点的寻找
随着甲基化研究水平的提高,近年来,提出了以限制性标记基因组扫描(RLGS)为代表的一系列新兴的全基因组甲基化扫描分析的新技术,另如:甲基化敏感的限制性指纹谱技术(methylation-sensitive restriction fingerprinting technique,MSRF)、甲基化间区位点扩增(amplification of inter-methylated sites,AIMS)等,这些新技术的出现为甲基化胚胎发育、肿瘤细胞异常基因印记向更深层次的发展提供了有效方法学工具。简单说来,[46][47][48] 1.RLGS是将稀有切点限制酶(NotⅠ)和二维凝胶电泳相结合,与NotⅠ-Eco RV文库进行对比分析,检测、扫描全基因组甲基化的情况。它的缺点是只能分析基因组中50%左右的CpG岛;2.MSRF用到限制性内切酶,如:MseⅠ、BstUⅠ,并采用10碱基随机引物扩增差异甲基化片段,几乎可以检测全所有CpG岛,但此方法复杂、需要后续鉴定、技术难度高;3.AIMS技术采用甲基化敏感和甲基化不敏感同裂酶(isoschizomer)裂解以及接头(adaptor)引物扩增甲基化间区序列,此方法可通过接头引物控制扩增带的复杂程度,且所得片段在200-2000bp之间,可以直接到载体并测序 。优点是简单方便,可以作为全差异印记基因筛选的有效工具。
2.3.1 限制性标记扫描(restriction landmark genomic scanning,RLGS)
Costello等2000年报道的RLGS[46,49]能对整个基因组的甲基化状态进行分析,发现新甲基化基因的方法。这种方法联合使用了限制性内切酶及二维电泳。其过程是:先用甲基化敏感的稀频限制性内切酶NotⅠ消化DNA ,甲基化位点保留,标记末端、切割、行一维电泳,随后再用更高频的甲基化不敏感的内切酶切割,行二维电泳,这样甲基化的部分被切割开并在电泳时显带,得到RLGS图谱与正常对照得出缺失条带即为甲基化的可能部位[5]。
这种方法可同时分析不同肿瘤中甲基化模式的异同和寻找肿瘤内DNA 甲基化的新靶点,由于新发现的甲基化新靶点的作用尚不清楚,因此其需要后续进一步分析确定,此外,RLGS图谱不能完全确认所缺失的片段是由于甲基化所致还是由于DNA 本身缺失所致[5]且结果分析复杂,不易解释。
2.3.2 MBD(Methyl-CpG binding domain column chromatography,甲基化结合区)柱层析法
根据MBD蛋白家族和MeCP2的特性,Masahiko Shiraishi等2004年[50]提出了一种新的方法MBD柱层析法,用于筛选和发现基因组中甲基化的情况。并且比较了MBD和重亚硫酸盐测序 法,得出:用MBD柱层析法分析得出的甲基化片段均能用重亚硫酸盐测序 法证实[50]。
现已较清楚CpG岛的甲基化在基因沉默中起着重要作用,但其过程中的很多具体环节及机制尚不清楚[51]。如:基因沉默是由启动子区个别位点CpG发生甲基化引起的,还是由全部的CpG发生甲基化而引起的。但研究认为启动子区胞嘧啶甲基化导致基因表达沉默并非是由于甲基基团阻碍了转录因子的结合,而是由于那些能与甲基化区特异性结合的蛋白发挥作用[52,53]而引起的。
现已发现一些能与CpG甲基化位点特异性结合的蛋白,一种是MeCP1,它能够与对称性多位点甲基化CpG位点相结合,另一种是MeCP2,不同于MeCP1的是它能够与单甲基化CpG位点特异结合,且不与半甲基化位点结合。而且报道的MBD1、MBD2、MBD3、MBD4蛋白都与MeCP2有着相同的特性[54,55]。
这种方法是:MBD柱中含有甲基化位点特异性结合蛋白的功能区(Methylation Binding Domain,MBD),能够与甲基化位点特异性结合。该蛋白一端通过连接多个组蛋白与凝胶结合,其另一端的多肽功能区暴露,这样当待测DNA 片段通过时,含有甲基化位点的DNA 即与MBD多肽牢固结合。在Masahiko Shiraishi等的研究中还发现,甲基化位点的数目是决定MBD柱结合力的最主要因素,而且,甲基化的密度也对其有重要的影响,也就是说,相同长度的DNA 片段中甲基化位点密度越高,其结合力就越强[50]。
这种方法的优点是:1.是一种高通量的检测方法;2.可对未知片段进行初筛,选出的含有甲基化的片段进一步检测以发现新的甲基化位点;3.方法快速、简便。缺点是:1.一些DNA 片段的特殊构型也可与MBD柱相结合,造成MBD非特异性捕获而引起假阳性(如:EcoRⅡ可与MBD结合);2.是一种定性而非定量的方法;3.由于MBD柱中所含多肽的量不完全相同,因此,使用不同的MBD柱或单个柱经多次使用得到的分离产物之间会存在差异[50]。
2.3.3 联合甲基化敏感性限制性内切酶的MBD(Combination of methylated-DNA precipitation and methylation-sensitive restriction enzymes,COMPARE-MS)
Srinivasan Yegnasubramanian 2006年[55]报道了一种新技术COMPARE-MS,该法将MBD柱层析法与MS-RE联用,互补了各自单用的弊处,能够快速、敏感的检测DNA 甲基化情况,可用于临床标本检测,作为早期诊断和肿瘤分级的依据[56]。其过程是:用非待测区的内切酶和甲基化敏感的限制性内切酶同时消化DNA 片段,随后通过MBD柱捕获,保留了含有甲基化区的片段,最后通过实时PCR扩增定量分析(见图9)。

图9:联合甲基化敏感性限制性内切酶的MBD(COMPARE-MS)示意图。用甲基化以外的位点的内切酶(A酶)与甲基化敏感的内切酶(B酶)联用,则甲基化的DNA 链不被切开,非甲基化的切开,再行MBD捕获存在甲基化位点的DNA 片段,最后行实时PCR扩增并分析;这就避免了因仅用内切酶(A酶+ B酶)而不用MBD捕获时由于内切酶消化不完全而引起假阳性的问题,同样避免了单用MBD时由于非特异性捕获而引起的假阳性(图9参考引文[55]并略加修改)。
这种方法联合运用了MBD柱层析法及MS-RE法,这就避免了单用MBD引起的非特异性捕获及MS-RE不完全消化引起的假阳性的问题。因此,在快速、简便检测的同时,亦保证了结果的特异性和敏感性。缺点是:需要使用限制性内切酶,因此不同程度地受到内切酶识别位点的限制。
研究甲基化的方法之多,从一个方面说明了甲基化研究难度之大,也从另一个方面说明这些方法都存在着一定的限制。面对具体问题,选择最合适的解决方法就显得尤为重要。首先,根据研究目的选择合适方法:是研究整体水平的甲基化还是特定位点的甲基化,或是要发现全中新的甲基化位点;其次,根据客观条件筛选方法,如:目标的序列是否已知,是定量研究还是定性研究,样本来源及数量如何,是否需要高通量的样本检测方法;最后,全面分析,选取敏感、可靠、经济、简便的方法,以达到理想的效果。随着甲基化研究的不断深入,甲基化分析技术将逐步完善。完善的研究技术将提供强有力的技术支持,从而为表观遗传、胚胎发育、基因印记及肿瘤研究提供一些新的思路。
参考文献 :
[1]Wu C T,Morris J R.Genes,genetics and epigenetics: a correspondence [J].Science,2001,293: 1103-1105.
[2]黄庆,郭颖,府伟灵.人类表观计划[J].生命的化学,2004,24(2): 101-102.
[3]Dahl C,Guldberg P.DNA methylation analysis techniques [J].Biogerontology,2003,4(4): 233-250.
[4]董玉玮,侯进慧,朱必才等.表观遗传学的相关概念和研究进展[J].生命的化学,2005,22(1): 1-3.
[5]武立鹏,朱卫国.DNA 甲基化的生物学应用及检测方法进展[J].中国检验医学杂志,2004,27(7): 468-474.
[6]Riggs A D,Jones P D.5-methylcytosine,gene regulation and cancer [J].Adv Cancer Res,1984,40: 1-30.
[7]Bird A P.CpG-rich islands and the function of DNA methylation [J].Nature,1986,321: 209-213.
[8]Cottrell S E.Molecular diagnostic applications of DNA methylation technology [J].Clin Biochem,2004,Jul,37(7): 595-604.
[9]Jones P A,Baylin S B.The fundamental role of epigenetic events in cancer [J].Nat Rev Genet,2002,Jun.3(6): 415-28.
[10]张永彪,褚嘉�.表观遗传学与人类疾病的研究进展 [J].遗传,2005,27(3): 466-472.
[11]Feinberg A P,Tycko B.The history of cancer epigenetic [J].Nat Rev Cancer,2004,4(2): 143-153.
[12]黄琼晓,金帆,黄荷凤.DNA 甲基化的研究方法学 [J].国外医学遗传学分册,2004,27(6): 354-358.
[13]Nuovo G J,Plaia T W,Belinsky S A,et al.In situ detection of the hypermethylation- induced inactivation of the p16 gene as an early event in oncogenesis [J].Proc Natl Acad Sci USA,1999,96: 12754-12759.
[14]Soares J,Pinto A E,Cunha C V,et al.Global DNA hypomethylation in breast carcinoma: correlation with prognostic factors and tumor ptogression [J].Cancer,1999,85: 112-118.
[15]Shinichi Toyooka,Nobuyoshi Shimizu.Models for studying DNA methylation in human cancer: a review of current status [J].Drug discovery today: Disease Model,2004,1(1): 37-42.
[16]Cui H,Horon I L,Ohlsson R,Hamilton S R,Feinberg A P.Loss of imprinting in normal tissue of colorectal cancer patients with microsatellite instability [J].Nat Med Nov,1998,4(11): 1276-80.
[17]Uhlmann K,Rohde K,Zeller C,et al.Distinct methylation profiles of glioma subtypes.[J] Int Cancer,2003,Aug.10,106(1): 52-59.
[18]Kuo K C,McCune R A,Gehrke C W,et al.Quantitative reversed-phase high performance liquid chromatographic determination of major and modified deoxyribonucleosides in DNA [J].Nucleic Acids Res,1980,8: 4763-4776.
[19]Fraga M F,Uriol E,Borja D L,et al.High-performance capillary electrophoretic method for the quantification of 5-methyl 2-deoxycytidine in genomic DNA : application to plant,animal and human cancer tissues [J].Electrophoresis,2002,23: 1677-1681.
[20]Oefner,P J,Bonn G K,Huber C G,et al.Comparative study of capillary zone electrophoresis and high-performance liquid chromatography in the analysis of oligonucleotides and DNA .[J].Chromatogr,1992,625(2): 331-3401.
[21]邓大君,邓国仁,吕有勇等.变性高效液相色谱 法检测CpG岛胞嘧啶甲基化 [J].中华医学杂志,2001,80(2),158-1611.
[22]Wu J,Issa J,Hermen J,et al.Expression of an exogenous eukaryotic DNA methyl transferase gene induces transformation of NIN3T3 ceils [J].Proc Natl Acad Sci USA,1993,90(19): 8891-8895.
[23]Oakeley E J,Podesta A,Jost J P.Developmental changes in DNA methylation of the two tobacco pollen nuclei during maturation [J].Proc Natl Acad Sci USA,1997,94: 11721-11725.
[24]Oakeley E J,Schmitt F,Jost J P.Quantification of 5-methylcytosine in DNA by the chloroacetaldehyde reaction [J].Biotechniques,1999,27: 744-6,748-50,752.
[25]Frommer M,McDonald L E,Millar D S,et al.A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands [J].Proc Natl Acad Sci USA,1992,89: 1827-1831.
[26]朱燕.DNA 的甲基化的分析与状态检测 [J].现代预防医学,2005,32(9): 1070-1073.
[27]沈佳尧,侯鹏,祭美菊等.DNA 甲基化方法研究现状 [J].生命的化学,2003,23(2): 149-151.
[28]Herman J G,Graff J R,Myohanen S,et al.Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands [J].Proc Natl Acad Sci USA,1996,Sep 3.93(18),9821-9826.
[29]Kuppuswamy M N,Hoffmann J W,Kasper C K,Spitzer S G,Groce S L,Bajaj S P.Single nucleotide primer extension to detect genetic diseases: experimental application to hemophilia B (factor IX)and cystic fibrosis genes [J].Proc Natl Acad Sci USA,1991,88: 1143-1147.
[30]Gonzalgo ml,Jones P A.Rapid quantitation of methylation differences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE)[J].Nucleic Acids Res,1997,25: 2529-2531.
[31]Xiong Z,Laird P W COBRA: a sensitive and quantitative DNA methylation assay [J].Nucleic Acids Res,1997,25: 2532-2534.
[32]Maekawa M,Sugano K,Kashiwabara H,et al.DNA
![十二烷基二甲基(3-磺丙基)氢氧化铵内盐 [用于生化研究],14933-08-5,≥98%,阿拉丁](https://img1.dxycdn.com/p/s14/2024/0619/475/6370229598169633081.jpg!wh200)



![5-溴-4-氯-3-吲哚基β-D-葡萄糖苷酸钠盐[用于生物学研究],129541-41-9,≥98%,阿拉丁](https://img1.dxycdn.com/p/s14/2024/0619/874/0460612821870833081.jpg!wh200)