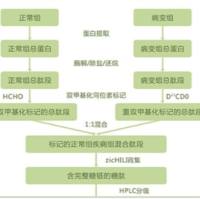
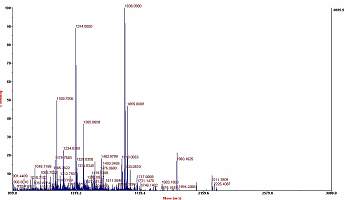
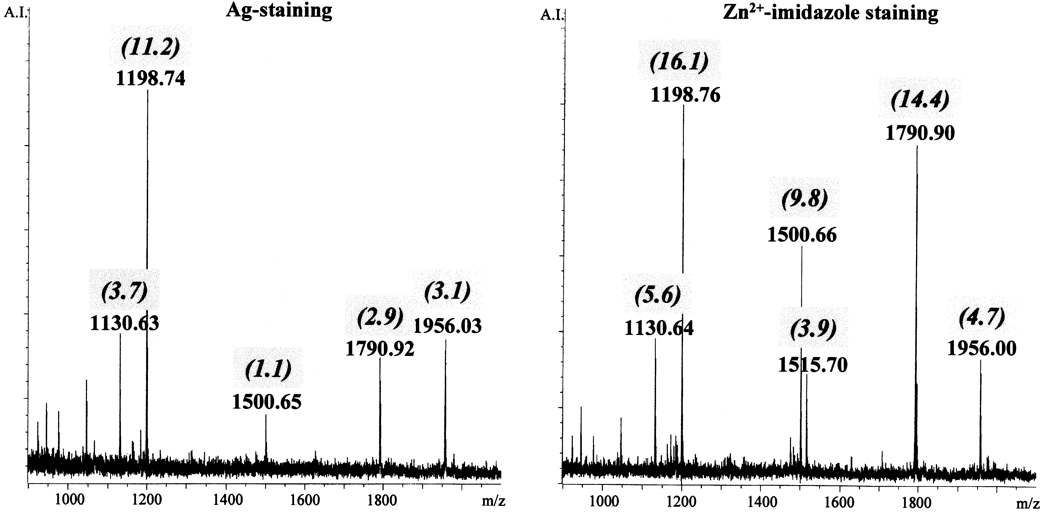
肽质量指纹图谱—MALDI-TOF 法鉴定蛋白质
丁香园
9077
1. 前言
值得注意的是,蛋白质质谱(MS) 鉴定是在 20 世纪 80 年代后期基于 “ 软” 电离技术发展起来的,这些电离技术包括欧洲 Michael Karas 和 Franz Hillenkamp 开发的基质辅助激光解吸电离(MALDI ) 和美国 John Fenn 开发的电喷雾电离(ESI )。JohnFenn 和 Takana 由于开发了 “软解吸电离方法应用于生物大分子的质谱分析” ,而获得了 2002 年诺贝尔化学奖。
由于蛋白质提取物能充分分离和兼容二维凝胶的染色(见第 16 章和第 17 章),因此适合于通过 MS 进行蛋白质鉴定。然而,整个蛋白质的 MS 不等于蛋白质的直接鉴定。要达到灵敏度高、准确度好并能访问大部分数据( 覆盖序列),蛋白质必须由内切蛋白酶(endoprotease) 消化产生多种蛋白质的特定片段。如果内切蛋白酶有足够的特异性、数据库中有蛋白质的信息,则通过对实验样品肽质量(由内切蛋白酶消化产生、通过 MALDI-TOF 质谱检测)与数据库中所有蛋白质硅片消化的肽质量的推断比较,就能对候选蛋白质进行鉴定。
植物蛋白质组学使用的技术类似于其他蛋白质组研究。然而,特别要注意样品生物起源的特殊性。事实上,与动物基因组相比,植物的基因组较大(通常为多倍体),目前只有少量的植物基因组已经完成了测序和注释。拟南芥是第一个完成基因组测序的植物(2000 年 12 月)。对于没有测序的植物,也许可以获得部分蛋白质信息;但对于大多数植物 [ 主要是木本和谷类(除了水稻)植物 ] 来说,基因组信息很少。因此对于没有测序的品种,也许可以通过同源性比较对蛋白质进行鉴定。对于未鉴定的蛋白质,MS/MS 肽测序可能是鉴定蛋白质的唯一有效方法(无论是 MALDI-TOF/TOF 还是纳米液相色谱法 [ ( LC ) -ESI-MS/MS] 。此外,不能首先使用植物特定的数据库,这是因为大部分蛋白质污染有人或哺乳动物的信息。
实验步骤如下所述。
( 1 ) 从凝胶上切除蛋白点,用胰蛋白酶进行凝胶消化(见 19.3.1)。
( 2 ) 将消化的样品沉积 MALDI 标基上,MALDI-TOF 谱获取信息并进行注释 ( 见 19.3.2)。
( 3 ) 数据库搜索,仔细查验搜索匹配的片段(见 19.3.3)。
2. 材料
2.1 仪器设备
( 1 ) 干净的真空离心机。
( 2 ) 干净的烤箱(高达 56°C )
( 3 ) 现代(20 世纪 90 年代末以来)MALDI-TOF 质谱仪(配有延迟或脉冲离子萃取技术和静电反射镜[反射 ( MALDI-reTOF) ] 。
2.2 试剂
1. 胶内消化
( 1 ) 水(HPLC 级或 MiUi-Q 级)。
( 2 ) 高纯度碳酸氢铵。
( 3 ) 乙腈 [ 高效液相色谱(HPLC) 级 ] 。
( 4 ) 三氟乙酸(TFA;HPLC 级)。
( 5 ) n-Octyl-glycopyranoside (n-OGP)。
( 6 ) 测序级猪胰蛋白酶。虽然也可用其他优质的测序级猪胰蛋白酶,但为了均一性和方便,使用 Promega 公司的测序级猪胰蛋白可以优化实验方案和数据。
( 7 ) 碳酸氢铵缓冲液:25 mmol/L 碳酸氢铵缓冲液,pH 7.8。
( 8 ) 乙腈/碳酸氢铵缓冲液:50/50 (V/V) 乙腈/25 mmol/L 碳酸氢铵缓冲液( pH 7.8)。
( 9 ) 消化缓冲液(冰浴配制):0.0125 μg/μl 测序级胰蛋白酶,溶解于 25 mmol/L 碳酸氢铵缓冲液(含 5 mmol/L n-OGP) 。
2. MALDI-TOF 质谱
( 1 ) α-氰基-4-羟基肉桂酸粉末,重结晶。
( 2 ) 水(HPLC 级或 Milli-Q 级)。
( 3 ) 乙腈(色谱纯)。
( 4 ) 丙酮(色谱纯 )。
( 5 ) 乙醇(色谱纯)。
( 6 ) TFA 溶液:0.1% TFA ( HPLC 级)水溶液。
( 7 ) 乙腈/ TFA 溶液:3 : 2(V/V)乙腈/ 0.1% TFA 水溶液。
( 8 ) 乙腈/ TFA 溶液:1 : 1 ( V/V ) 乙腈/0.1% TFA 水溶液。
( 9 ) 乙醇/丙酮/TFA 溶液:6 : 3 : 1 ( V/V/V ) 乙醇/丙酮/0.1% TFA 水溶液。
1. 注释
( 1 ) 自动化切胶、胶内酶解消化和通过特异液相处理收集目的样品。为了提高通量效率,大规模蛋白质组学设备应该配备自动化处理收集技术,以减少样品在处理过程中受人类角蛋白污染的可能性。
( 2 ) 切取蛋白质点面积过大不利于蛋白质的鉴定。因为面积过大会增加化学噪音背景,降低胰蛋白酶在蛋白点中的渗透能力,不利于蛋白酶扩散到蛋白点凝胶中。
( 3 ) 双向电泳中,在进行 SDS-PAGE 之前的平衡过程中蛋白质被还原(一般采用 DTT 或 β-巯基乙醇)和烷基化(一般采用碘乙酰胺或碘乙酸)。这样打断了半胱氨酸残基之间的二硫键,从而可防止蛋白质形成二级和三级结构来干扰蛋白质在聚丙烯酰胺凝胶中的迁移。对于一向电泳分离的蛋白质而言,在胶内酶解之前,必须进行还原和烷基化 (见第 22 章 ) 。
( 4 ) 由于胰蛋白酶的独特性,使它成为在蛋白质胶内酶解之后用于 MS 鉴定的合适内切酶:
a. 分子质量相对低的内切酶(约 24 kDa) ,用水化缓冲液溶解之后很容易渗透进入冷冻干燥的含蛋白质点的胶粒中。
b. 优良的酶切特性(酶切 K 和 R 的 C 端,如果下一个氨基酸是 P 则不进行酶切,如果 D/E 氨基酸的支链酸性端出现在旁边,则它的酶切效率降低,由于支链的空间效应,如果大量 F/W/Y 氨基酸出现在旁边,则它的酶切效率降低)。商业猪胰蛋白酶用 N-甲苯磺酰-L-苯基丙氨酸氯甲基酮(TPCK ) 进行了处理从而避免非特异性胰蛋白酶的活性。
c. 在分子质量 600~4000 Da 中,由于氨基酸 K 和 R 的存在,通过胰蛋白酶作用,产生了大部分的蛋白酶水解肽段,这和高分子质量精确的 MALDI-TOF-MS 兼容。
d. 商业猪胰蛋白酶赖氨酸侧链进行了甲基化还原处理,这样可减少自切肽段,但是在 842.5099 Th 和 2211.1046 Th 的两个信号强的离子峰能用于质谱内的校正平衡。
e. 蛋白酶水解肽段很容易就能质子化(并且在质谱分析中产生很好的信号),是因为肽段的两个基本位点,一个位于肽段 N 端,一个位于最后 C 端氨基酸( K 或 R ) 的支链上。此外,这个特性在 ESI 质谱中非常有用,通过 MS/MS 能够得到主要双电荷形式,从而产生有用的 b/y 离子片段系列(确定的电荷片段:见第 25 章 )。
( 5 ) 对于特需目的(如为了增加蛋白质的覆盖率),采用同样的方案,也可以使用其他蛋白内切酶。
a. Chrymotrypsin 在 F/W/Y 后进行酶切,产生更多、更短的片段。
b. Endo Lys-C 在 K 后进行酶切(对 R 不进行酶切)。
c. 蛋白酶 V8 在 DE 或 E 后进行酶切(取决于酶切消化缓冲液的 pH 范围)。
( 6 ) 推荐在冰上配置胰蛋白酶酶液并且放置在冰上使酶液充分渗透进入蛋白质胶粒中,这样可减少胰蛋白酶的自切作用。
( 7 ) 在 37°C 下酶解消化 3 h 以上,以过夜为最长酶解时间。过夜消化最为方便。对于长时间消化(如过夜),酶切消化过程可以直接在室温下进行而不用放在 37°C 的环境中。这样可以减少胰蛋白酶自切峰产生的信号。
( 8 ) 在干燥胶粒的方法中,其目的是为了获得均匀的小结晶。长时间结晶会产生大结晶,相反快速结晶会得到较小的结晶。为了掌握结晶过程并且不改变其组分,胶粒可在真空状态或温箱中加热(30~40°C ) 干燥几分钟。
( 9 ) 一些 MOLDI-TOF 质谱仪的制造商已经开发出了预制的疏水/亲水点样板(如 Bruker Daltonics 的 AnchorChipTM 技术)。这些点样板能使样品精确放置并且在点样板上进行浓缩,因此离子信号强度的灵敏性能提高 5~20 倍 。同时认真仔细清洗点样板很重要,这样能防止外来污染(主要是塑化剂引起的污染)浓缩在点样孔中。胶内酶解后的肽段可以直接在点样板上点样( 见 3.1.1 节),或通过肽段抽提和浓缩后再点样。
( 10 ) 从质谱分析的角度看,能否得到可信的蛋白质鉴定结果有两点很重要:
a. 质量的精确度 [ 理想的情况应低于 30 ppm ( 1 ppm=10-6,后同),通过仔细认真的光谱内参校正,如今的 MALDI-TOF 质谱仪已经能达到 10~15 ppm] 。
b. 检测尽可能多的离子灵敏性来增加序列覆盖率。
( 11 ) 通过精确的运算法则,光谱的注释一般是自动完成的。但是如果蛋白质的鉴定是失败的,那么应该对注释进行检查并修改它到:
a. 如果双向电泳的蛋白点含有一个或两个可鉴定的蛋白质,最多选择 40 个信号最强的离子峰(如两个蛋白点具有相近的表达模式)。
b. 最小的信噪比为 3 : 1。
( 12 ) 对于信号强的质量光谱,低信号的离子注释会降低蛋白质的鉴定率,因此在大多数强信号蛋白质特异肽段离子中,离子信号低于 2% 的应排除。
( 13 ) Promega 公司生产的测序级猪胰蛋白酶的主要已知自解峰是:842. 51、870. 54、1045.56、1940. 94、2211.10、2225.12、2239.14、2283.18、2299.18 和 2807.31。在网站 http: //prospector, ucsf. edu /ucsfhtml4. 0/misc/trypsiru htm 上可以查询到完整的姨蛋白酶自解峰。
( 14 ) 我们推荐对已知的污染物同时进行注释(如非植物角蛋白峰),在第一步数据库搜索步骤中同时对这些污染物进行注释,然后精确鉴定这些污染物后再过滤这些污染物所产生的峰值。事实上,角蛋白水解峰(如 1475.74 Th ) 可能有不同的角蛋白亚类 ( 如角蛋白 10 和角蛋白 2) ,其他角蛋白水解检测到的特异峰属于这些亚类中的一类或几类。
( 15 ) 室内授权的搜索引擎(如 Mascot):
a. 允许特异或所有蛋白质数据库的安装。
b. 保留一些研究所需的机密性。
c. 搜索时间不受网速和远程服务器忙碌程度的限制。
d. 允许批量搜索。
e. 允许所有搜索结果没有时间限制进行存档。
( 16 ) 搜索引擎网址链接:
a. Mascot:http://www.matrixscience. com/cgi/search _ form, pi? FORMVER= 2 & S E A R C H = P M F
b. MS-Fit (protein prospector) : http: // prospector, ucsf. edu/ucsfhtml4. 0/msfit. htm
c. ProFound:http : //prowl, rockefeller, edu/profound _ b i n / W e b P r o F o u n d . exe
d. Peptide search : http: // w w w . m a n n . e m b l - h e i d e l b e r g . d e / G r o u p P a g e s /PageLink/peptidesearch/Services/PeptideSearch/FR _PeptideSearchFormG4. html
e. Pep MAPPER:http:// wolf. bms. umist. ac. u k / m a p p e r /
f. Aldente: http:// www . expasy. org/ tools/aldente
( 17 ) 单独的序列覆盖信息理解起来有局限性,它还部分依赖于预测蛋白质的分子质量。对于一个高分子质量的预测蛋白质而言,低序列覆盖度可能表明:
a. 胰蛋白酶无法完全进入此蛋白质的某些部分。
b. 此蛋白质包含很少的胰蛋白酶酶切位点 [ 如富含脯氨酸蛋白(prolinerich proteins,PRP)], 所以酶切肽段过大以至于质谱分析中无法检测到 (超出了质量检测范围 )。
c. 肽段离子化竞争效应使某些肽段无法离子化。
d. 用于胶内酶解的蛋白点仅是这个预测蛋白质的一部分。
e. 所搜索的蛋白质数据库中没有准确包含胶内酶解蛋白点的序列(突变或翻译后修饰,但是更典型的序列多态性的原因是种间交叉)。
另一方面,对于低分子质量的预测蛋白质,髙序列覆盖率可能就完全表明在数据库中记录的此蛋白序列太短,从而导致序列覆盖率不正常地过高。
对于这些原因,通过对匹配肽段在蛋白质序列中的位置进行再次比对确认要比要比序列覆盖率合适得多:匹配肽段是否从头到尾都位于蛋白质序列上( 这可能表明预测的有效性)?或者相反,它们的部分序列位于蛋白质序列上( 如果它与胶内分子质量和等电点一致,这可能表明仅鉴定到了一个蛋白质降解片段)?
( 18 ) 在单向凝胶电泳中,多个并联蛋白质可能在挖胶过程中同时被挖取出来。如果发生这种情况,那么我们必须选择合适的数据库进行搜索(见第 22 章)。
( 19 ) 大量的可变修饰搜索参数使搜索时间延长并会产生非特异性蛋白质鉴定结果 。如果这些可变修饰搜索参数对于搜索过程是必要的,我们推荐限制其他搜索参数(如较低的质量误差、对物种的分类进行更精确的描述和设置丢失的酶切位点数目不存在)。
参考文献
1. Karas M., B a c h m a n D., Bahr U., and Hillenkamp F. (1987) Matrix-assisted ultraviolet laser desorption of non-volatile compounds. Int. J. Mass Spectrom. IonProc.53-68.
2. Karas M . and Hillenkamp F. (1988) Laser desorption ionization of proteins withmolecular masses exceeding 10,000 daltons. Anal. Chem.2299-2301.
3. Karas M., Bahr U., Ingendoh A., and Hillenkamp F. (1989) Laser desorption/ionisation mass spectrometry of proteins of mass 100,000 to 250,000 Dalton.Angewandte Chemie Int. Ed.760-761.
4. Fenn J. B., M a n n M., M e n g C. K., W o n g S. F., and Whitehouse C. M . (1989)Electrospray ionization for mass spectrometry of large biomolecules. Science64— 71.
5. http://nobelprize.org/chemistry/laureates/2002/index.html .
6. Tanaka K., W a k i H., Ido Y., Akita S., Yoshida Y., and Yoshida T. (1988) Proteinand polymer analysis up to m/z 100,000 by laser ionisation time-of-flight massspectrometry. Rapid Comm. Mass Spectrom.151-153.
7. ArabidopsisG e n o m e Initiative (2000) Analysis of the g e n o m e sequence of theflowering plant. Nature796-815.
8. Jensen O., W i l m M., Schevschenko A., and M a n n M . (1999) Sample preparationmethods for mass spectrometric peptide mapping directly from 2 - D E gels, inMethods in Molecular Biology: 2-D Proteome Analysis Protocols,vol. 112 (Link,A. J., ed.), H u m a n a , Totowa, NJ, pp. 513-530.
9. D e Hoffmann, E., Charette, J., and Stroobant, V. (1999) Spectrometrie de Masse,Cours et Exercices Corriges,2 e m e edition. Dunod, Paris.
10. D e Hoffmann, E. and Stroobant, V. (2002) Mass Spectrometry: Principles andApplications,2nd ed. John Wiley & Sons, Hoboken, NJ.
11. Throck Watson, J. (1997) Introduction to Mass Spectrometry,3rd ed. Lippincott-Raven: N e w York.
12. James, P. (ed.) (2000) Proteome Research: Mass Spectrometry (Principles andPractice).Springer Verlag, N e w York.
13. Cotter, R. J. (1997) Time-of-Flight Mass Spectrometry: Instrumentation andApplications in Biological Research.American Chemical Society, Washington, DC.
14. Vestal M . L., Juhasz P., Hines W., and Martin S. (1998) in Proceedings of the46th ASMS Conference on Mass Spectrometry and Allied Topics,Orlando, FL.
值得注意的是,蛋白质质谱(MS) 鉴定是在 20 世纪 80 年代后期基于 “ 软” 电离技术发展起来的,这些电离技术包括欧洲 Michael Karas 和 Franz Hillenkamp 开发的基质辅助激光解吸电离(MALDI ) 和美国 John Fenn 开发的电喷雾电离(ESI )。JohnFenn 和 Takana 由于开发了 “软解吸电离方法应用于生物大分子的质谱分析” ,而获得了 2002 年诺贝尔化学奖。
由于蛋白质提取物能充分分离和兼容二维凝胶的染色(见第 16 章和第 17 章),因此适合于通过 MS 进行蛋白质鉴定。然而,整个蛋白质的 MS 不等于蛋白质的直接鉴定。要达到灵敏度高、准确度好并能访问大部分数据( 覆盖序列),蛋白质必须由内切蛋白酶(endoprotease) 消化产生多种蛋白质的特定片段。如果内切蛋白酶有足够的特异性、数据库中有蛋白质的信息,则通过对实验样品肽质量(由内切蛋白酶消化产生、通过 MALDI-TOF 质谱检测)与数据库中所有蛋白质硅片消化的肽质量的推断比较,就能对候选蛋白质进行鉴定。
植物蛋白质组学使用的技术类似于其他蛋白质组研究。然而,特别要注意样品生物起源的特殊性。事实上,与动物基因组相比,植物的基因组较大(通常为多倍体),目前只有少量的植物基因组已经完成了测序和注释。拟南芥是第一个完成基因组测序的植物(2000 年 12 月)。对于没有测序的植物,也许可以获得部分蛋白质信息;但对于大多数植物 [ 主要是木本和谷类(除了水稻)植物 ] 来说,基因组信息很少。因此对于没有测序的品种,也许可以通过同源性比较对蛋白质进行鉴定。对于未鉴定的蛋白质,MS/MS 肽测序可能是鉴定蛋白质的唯一有效方法(无论是 MALDI-TOF/TOF 还是纳米液相色谱法 [ ( LC ) -ESI-MS/MS] 。此外,不能首先使用植物特定的数据库,这是因为大部分蛋白质污染有人或哺乳动物的信息。
实验步骤如下所述。
( 1 ) 从凝胶上切除蛋白点,用胰蛋白酶进行凝胶消化(见 19.3.1)。
( 2 ) 将消化的样品沉积 MALDI 标基上,MALDI-TOF 谱获取信息并进行注释 ( 见 19.3.2)。
( 3 ) 数据库搜索,仔细查验搜索匹配的片段(见 19.3.3)。
2. 材料
2.1 仪器设备
( 1 ) 干净的真空离心机。
( 2 ) 干净的烤箱(高达 56°C )
( 3 ) 现代(20 世纪 90 年代末以来)MALDI-TOF 质谱仪(配有延迟或脉冲离子萃取技术和静电反射镜[反射 ( MALDI-reTOF) ] 。
2.2 试剂
1. 胶内消化
( 1 ) 水(HPLC 级或 MiUi-Q 级)。
( 2 ) 高纯度碳酸氢铵。
( 3 ) 乙腈 [ 高效液相色谱(HPLC) 级 ] 。
( 4 ) 三氟乙酸(TFA;HPLC 级)。
( 5 ) n-Octyl-glycopyranoside (n-OGP)。
( 6 ) 测序级猪胰蛋白酶。虽然也可用其他优质的测序级猪胰蛋白酶,但为了均一性和方便,使用 Promega 公司的测序级猪胰蛋白可以优化实验方案和数据。
( 7 ) 碳酸氢铵缓冲液:25 mmol/L 碳酸氢铵缓冲液,pH 7.8。
( 8 ) 乙腈/碳酸氢铵缓冲液:50/50 (V/V) 乙腈/25 mmol/L 碳酸氢铵缓冲液( pH 7.8)。
( 9 ) 消化缓冲液(冰浴配制):0.0125 μg/μl 测序级胰蛋白酶,溶解于 25 mmol/L 碳酸氢铵缓冲液(含 5 mmol/L n-OGP) 。
2. MALDI-TOF 质谱
( 1 ) α-氰基-4-羟基肉桂酸粉末,重结晶。
( 2 ) 水(HPLC 级或 Milli-Q 级)。
( 3 ) 乙腈(色谱纯)。
( 4 ) 丙酮(色谱纯 )。
( 5 ) 乙醇(色谱纯)。
( 6 ) TFA 溶液:0.1% TFA ( HPLC 级)水溶液。
( 7 ) 乙腈/ TFA 溶液:3 : 2(V/V)乙腈/ 0.1% TFA 水溶液。
( 8 ) 乙腈/ TFA 溶液:1 : 1 ( V/V ) 乙腈/0.1% TFA 水溶液。
( 9 ) 乙醇/丙酮/TFA 溶液:6 : 3 : 1 ( V/V/V ) 乙醇/丙酮/0.1% TFA 水溶液。
1. 注释
( 1 ) 自动化切胶、胶内酶解消化和通过特异液相处理收集目的样品。为了提高通量效率,大规模蛋白质组学设备应该配备自动化处理收集技术,以减少样品在处理过程中受人类角蛋白污染的可能性。
( 2 ) 切取蛋白质点面积过大不利于蛋白质的鉴定。因为面积过大会增加化学噪音背景,降低胰蛋白酶在蛋白点中的渗透能力,不利于蛋白酶扩散到蛋白点凝胶中。
( 3 ) 双向电泳中,在进行 SDS-PAGE 之前的平衡过程中蛋白质被还原(一般采用 DTT 或 β-巯基乙醇)和烷基化(一般采用碘乙酰胺或碘乙酸)。这样打断了半胱氨酸残基之间的二硫键,从而可防止蛋白质形成二级和三级结构来干扰蛋白质在聚丙烯酰胺凝胶中的迁移。对于一向电泳分离的蛋白质而言,在胶内酶解之前,必须进行还原和烷基化 (见第 22 章 ) 。
( 4 ) 由于胰蛋白酶的独特性,使它成为在蛋白质胶内酶解之后用于 MS 鉴定的合适内切酶:
a. 分子质量相对低的内切酶(约 24 kDa) ,用水化缓冲液溶解之后很容易渗透进入冷冻干燥的含蛋白质点的胶粒中。
b. 优良的酶切特性(酶切 K 和 R 的 C 端,如果下一个氨基酸是 P 则不进行酶切,如果 D/E 氨基酸的支链酸性端出现在旁边,则它的酶切效率降低,由于支链的空间效应,如果大量 F/W/Y 氨基酸出现在旁边,则它的酶切效率降低)。商业猪胰蛋白酶用 N-甲苯磺酰-L-苯基丙氨酸氯甲基酮(TPCK ) 进行了处理从而避免非特异性胰蛋白酶的活性。
c. 在分子质量 600~4000 Da 中,由于氨基酸 K 和 R 的存在,通过胰蛋白酶作用,产生了大部分的蛋白酶水解肽段,这和高分子质量精确的 MALDI-TOF-MS 兼容。
d. 商业猪胰蛋白酶赖氨酸侧链进行了甲基化还原处理,这样可减少自切肽段,但是在 842.5099 Th 和 2211.1046 Th 的两个信号强的离子峰能用于质谱内的校正平衡。
e. 蛋白酶水解肽段很容易就能质子化(并且在质谱分析中产生很好的信号),是因为肽段的两个基本位点,一个位于肽段 N 端,一个位于最后 C 端氨基酸( K 或 R ) 的支链上。此外,这个特性在 ESI 质谱中非常有用,通过 MS/MS 能够得到主要双电荷形式,从而产生有用的 b/y 离子片段系列(确定的电荷片段:见第 25 章 )。
( 5 ) 对于特需目的(如为了增加蛋白质的覆盖率),采用同样的方案,也可以使用其他蛋白内切酶。
a. Chrymotrypsin 在 F/W/Y 后进行酶切,产生更多、更短的片段。
b. Endo Lys-C 在 K 后进行酶切(对 R 不进行酶切)。
c. 蛋白酶 V8 在 DE 或 E 后进行酶切(取决于酶切消化缓冲液的 pH 范围)。
( 6 ) 推荐在冰上配置胰蛋白酶酶液并且放置在冰上使酶液充分渗透进入蛋白质胶粒中,这样可减少胰蛋白酶的自切作用。
( 7 ) 在 37°C 下酶解消化 3 h 以上,以过夜为最长酶解时间。过夜消化最为方便。对于长时间消化(如过夜),酶切消化过程可以直接在室温下进行而不用放在 37°C 的环境中。这样可以减少胰蛋白酶自切峰产生的信号。
( 8 ) 在干燥胶粒的方法中,其目的是为了获得均匀的小结晶。长时间结晶会产生大结晶,相反快速结晶会得到较小的结晶。为了掌握结晶过程并且不改变其组分,胶粒可在真空状态或温箱中加热(30~40°C ) 干燥几分钟。
( 9 ) 一些 MOLDI-TOF 质谱仪的制造商已经开发出了预制的疏水/亲水点样板(如 Bruker Daltonics 的 AnchorChipTM 技术)。这些点样板能使样品精确放置并且在点样板上进行浓缩,因此离子信号强度的灵敏性能提高 5~20 倍 。同时认真仔细清洗点样板很重要,这样能防止外来污染(主要是塑化剂引起的污染)浓缩在点样孔中。胶内酶解后的肽段可以直接在点样板上点样( 见 3.1.1 节),或通过肽段抽提和浓缩后再点样。
( 10 ) 从质谱分析的角度看,能否得到可信的蛋白质鉴定结果有两点很重要:
a. 质量的精确度 [ 理想的情况应低于 30 ppm ( 1 ppm=10-6,后同),通过仔细认真的光谱内参校正,如今的 MALDI-TOF 质谱仪已经能达到 10~15 ppm] 。
b. 检测尽可能多的离子灵敏性来增加序列覆盖率。
( 11 ) 通过精确的运算法则,光谱的注释一般是自动完成的。但是如果蛋白质的鉴定是失败的,那么应该对注释进行检查并修改它到:
a. 如果双向电泳的蛋白点含有一个或两个可鉴定的蛋白质,最多选择 40 个信号最强的离子峰(如两个蛋白点具有相近的表达模式)。
b. 最小的信噪比为 3 : 1。
( 12 ) 对于信号强的质量光谱,低信号的离子注释会降低蛋白质的鉴定率,因此在大多数强信号蛋白质特异肽段离子中,离子信号低于 2% 的应排除。
( 13 ) Promega 公司生产的测序级猪胰蛋白酶的主要已知自解峰是:842. 51、870. 54、1045.56、1940. 94、2211.10、2225.12、2239.14、2283.18、2299.18 和 2807.31。在网站 http: //prospector, ucsf. edu /ucsfhtml4. 0/misc/trypsiru htm 上可以查询到完整的姨蛋白酶自解峰。
( 14 ) 我们推荐对已知的污染物同时进行注释(如非植物角蛋白峰),在第一步数据库搜索步骤中同时对这些污染物进行注释,然后精确鉴定这些污染物后再过滤这些污染物所产生的峰值。事实上,角蛋白水解峰(如 1475.74 Th ) 可能有不同的角蛋白亚类 ( 如角蛋白 10 和角蛋白 2) ,其他角蛋白水解检测到的特异峰属于这些亚类中的一类或几类。
( 15 ) 室内授权的搜索引擎(如 Mascot):
a. 允许特异或所有蛋白质数据库的安装。
b. 保留一些研究所需的机密性。
c. 搜索时间不受网速和远程服务器忙碌程度的限制。
d. 允许批量搜索。
e. 允许所有搜索结果没有时间限制进行存档。
( 16 ) 搜索引擎网址链接:
a. Mascot:http://www.matrixscience. com/cgi/search _ form, pi? FORMVER= 2 & S E A R C H = P M F
b. MS-Fit (protein prospector) : http: // prospector, ucsf. edu/ucsfhtml4. 0/msfit. htm
c. ProFound:http : //prowl, rockefeller, edu/profound _ b i n / W e b P r o F o u n d . exe
d. Peptide search : http: // w w w . m a n n . e m b l - h e i d e l b e r g . d e / G r o u p P a g e s /PageLink/peptidesearch/Services/PeptideSearch/FR _PeptideSearchFormG4. html
e. Pep MAPPER:http:// wolf. bms. umist. ac. u k / m a p p e r /
f. Aldente: http:// www . expasy. org/ tools/aldente
( 17 ) 单独的序列覆盖信息理解起来有局限性,它还部分依赖于预测蛋白质的分子质量。对于一个高分子质量的预测蛋白质而言,低序列覆盖度可能表明:
a. 胰蛋白酶无法完全进入此蛋白质的某些部分。
b. 此蛋白质包含很少的胰蛋白酶酶切位点 [ 如富含脯氨酸蛋白(prolinerich proteins,PRP)], 所以酶切肽段过大以至于质谱分析中无法检测到 (超出了质量检测范围 )。
c. 肽段离子化竞争效应使某些肽段无法离子化。
d. 用于胶内酶解的蛋白点仅是这个预测蛋白质的一部分。
e. 所搜索的蛋白质数据库中没有准确包含胶内酶解蛋白点的序列(突变或翻译后修饰,但是更典型的序列多态性的原因是种间交叉)。
另一方面,对于低分子质量的预测蛋白质,髙序列覆盖率可能就完全表明在数据库中记录的此蛋白序列太短,从而导致序列覆盖率不正常地过高。
对于这些原因,通过对匹配肽段在蛋白质序列中的位置进行再次比对确认要比要比序列覆盖率合适得多:匹配肽段是否从头到尾都位于蛋白质序列上( 这可能表明预测的有效性)?或者相反,它们的部分序列位于蛋白质序列上( 如果它与胶内分子质量和等电点一致,这可能表明仅鉴定到了一个蛋白质降解片段)?
( 18 ) 在单向凝胶电泳中,多个并联蛋白质可能在挖胶过程中同时被挖取出来。如果发生这种情况,那么我们必须选择合适的数据库进行搜索(见第 22 章)。
( 19 ) 大量的可变修饰搜索参数使搜索时间延长并会产生非特异性蛋白质鉴定结果 。如果这些可变修饰搜索参数对于搜索过程是必要的,我们推荐限制其他搜索参数(如较低的质量误差、对物种的分类进行更精确的描述和设置丢失的酶切位点数目不存在)。
参考文献
1. Karas M., B a c h m a n D., Bahr U., and Hillenkamp F. (1987) Matrix-assisted ultraviolet laser desorption of non-volatile compounds. Int. J. Mass Spectrom. IonProc.53-68.
2. Karas M . and Hillenkamp F. (1988) Laser desorption ionization of proteins withmolecular masses exceeding 10,000 daltons. Anal. Chem.2299-2301.
3. Karas M., Bahr U., Ingendoh A., and Hillenkamp F. (1989) Laser desorption/ionisation mass spectrometry of proteins of mass 100,000 to 250,000 Dalton.Angewandte Chemie Int. Ed.760-761.
4. Fenn J. B., M a n n M., M e n g C. K., W o n g S. F., and Whitehouse C. M . (1989)Electrospray ionization for mass spectrometry of large biomolecules. Science64— 71.
5. http://nobelprize.org/chemistry/laureates/2002/index.html .
6. Tanaka K., W a k i H., Ido Y., Akita S., Yoshida Y., and Yoshida T. (1988) Proteinand polymer analysis up to m/z 100,000 by laser ionisation time-of-flight massspectrometry. Rapid Comm. Mass Spectrom.151-153.
7. ArabidopsisG e n o m e Initiative (2000) Analysis of the g e n o m e sequence of theflowering plant. Nature796-815.
8. Jensen O., W i l m M., Schevschenko A., and M a n n M . (1999) Sample preparationmethods for mass spectrometric peptide mapping directly from 2 - D E gels, inMethods in Molecular Biology: 2-D Proteome Analysis Protocols,vol. 112 (Link,A. J., ed.), H u m a n a , Totowa, NJ, pp. 513-530.
9. D e Hoffmann, E., Charette, J., and Stroobant, V. (1999) Spectrometrie de Masse,Cours et Exercices Corriges,2 e m e edition. Dunod, Paris.
10. D e Hoffmann, E. and Stroobant, V. (2002) Mass Spectrometry: Principles andApplications,2nd ed. John Wiley & Sons, Hoboken, NJ.
11. Throck Watson, J. (1997) Introduction to Mass Spectrometry,3rd ed. Lippincott-Raven: N e w York.
12. James, P. (ed.) (2000) Proteome Research: Mass Spectrometry (Principles andPractice).Springer Verlag, N e w York.
13. Cotter, R. J. (1997) Time-of-Flight Mass Spectrometry: Instrumentation andApplications in Biological Research.American Chemical Society, Washington, DC.
14. Vestal M . L., Juhasz P., Hines W., and Martin S. (1998) in Proceedings of the46th ASMS Conference on Mass Spectrometry and Allied Topics,Orlando, FL.