文献解读:纽约市废水系统宏病毒组初探
&深圳市易基因科技有限公司
研究简介
噬菌体是迄今为止所研究的所有微生物群落中大量的成员,通过与其细菌宿主的相互作用影响微生物群落。尽管噬菌体在功能上很重要,而且无处不在,但与细菌相比,它们在城市环境中的探索还不够充分。废水是微生物生命的丰富来源,含有细菌、病毒和其他微生物,这些微生物存在于人类排泄物和环境径流源中。
2020年6月16日,纽约大学基因组学与系统生物学中心E. Ghedin研究团队在mSystems发表了题为Initial Mapping of the New York City Wastewater Virome的研究论文,文章描述了纽约市所有五个行政区的污水样本的病毒群落,并发现各采样点有独特的病毒群落特征。研究集中在细菌噬菌体(细菌病毒)对整个微生态系统的影响。并且发现了几个新的噬菌体簇,并成功地将它们与细菌宿主相关联,这为研究城市污水中的病毒-宿主相互作用提供了新的视角。该研究为首次研究了纽约市污水系统中存在的病毒群落,并指出了它们在纽约废水环境中的功能重要性。
研究结果:
本研究主要针对纽约市五个行政区的14个污水处理中心(图1)的16个样本点(于2014年11月取样)的宏基因组数据集进行宏病毒组学分析,平均每个样本10.5M双端reads(PE250)。利用VirMAP共确定16个样本总序列数的4.1%为病毒序列(6,993,448条reads),鉴定获得806种病毒分类注释结果。16个样本中平均单样本鉴定166类病毒(范围从78到480)。鉴定到的总病毒序列中约有99.9%注释为细菌噬菌体,剩余的极少数注释为真核病毒。需要指出的是鉴定的病毒序列大概90%仅仅注释到Virus级别(taxId=10239)。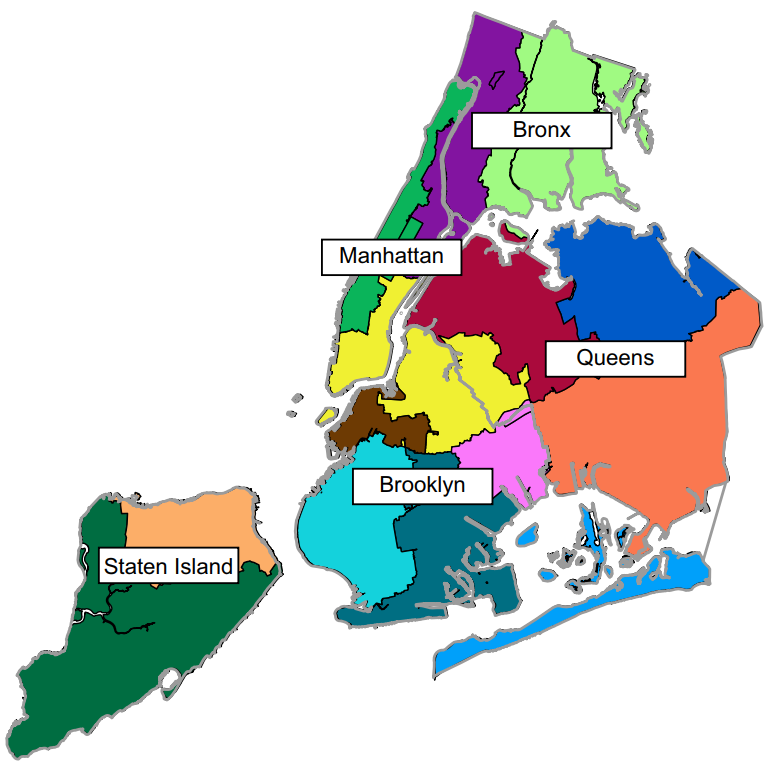
图1:纽约市污水处理系统及样本点。
为比较样本及位点之间病毒物种组成多样性的差异,计算了每个样本的α多样性指数Gini-Simpson’s Index (1-Simpson’s Index)。发现样本的多样性指数都很高(平均0.9)但物种分布并不均匀,表明样本中所包含的物种种类比较简单。利用Bray-Curtis算法计算样本之间β多样性指数,平均β多样性得分为0.04,表明样本之间有着相似的物种组成模式。换言之,取样位点的差异并没有显著影响到样本的物种组成分布(图2a)。为进一步判断不同取样点间病毒群落组成的差异,绘制upset图发现所有样本的核心病毒组物种数目为38,Queens区域独有的病毒物种数目最多为249(图2b)。选取丰度top20的病毒物种进行热图展示,发现高丰度的病毒主要包括Siphoviridae phages(Lactococcus phage 1706和Enterobacter phage phiEap-2); Myoviridae phages(Vibrio phage VH7D 和Shewanella sp. phage 3/49); Podoviridae phages(Acinetobacter phage Presley和Cronobacter phage vB_CsaP_Ss1 (图2c)。
图2:病毒组注释及多样性分析
为进一步探索在严格依赖于数据库比对方法之外是否有新的病毒序列可以鉴定出来,研究团队利用VirSorter软件对组装后的contigs进行了预测。在所有样本的contigs中共鉴定出来4881条病毒contigs。紧接着,作者利用基因网络分析将预测的contigs聚类为病毒簇(VCs),并通过可视化网络展现预测的病毒contigs与RefSeq数据库中已知的细菌和古菌病毒之间的关系(图3)。最终发现所有样本中仅4%的病毒contigs与RefSeq中的病毒基因组聚类成功。从图中可知,所有样本中都发现至少有一条contigs与最近鉴定的人类肠道中的crAssphage基因组聚类关联。有一半的样本中均有病毒contigs与水环境典型噬菌体Flavobacterium phage 11b基因组聚类关联。同时不同街区的病毒contigs也发现一些特殊性。例如,两个Queens街区样本(9和15)都有病毒contigs与Rhodobacter phage RcRhea聚集在一起,而该噬菌体已知能感染光合作用细菌 Rhodobacter capsulatus。Brooklyn街区的两个样本(4和5)聚集了Achromobacter phage JWF,该噬菌体最近被报道从污水中分离出来,能感染一种潮湿环境中新的院内感染病原菌Achromobacter xylosoxidans。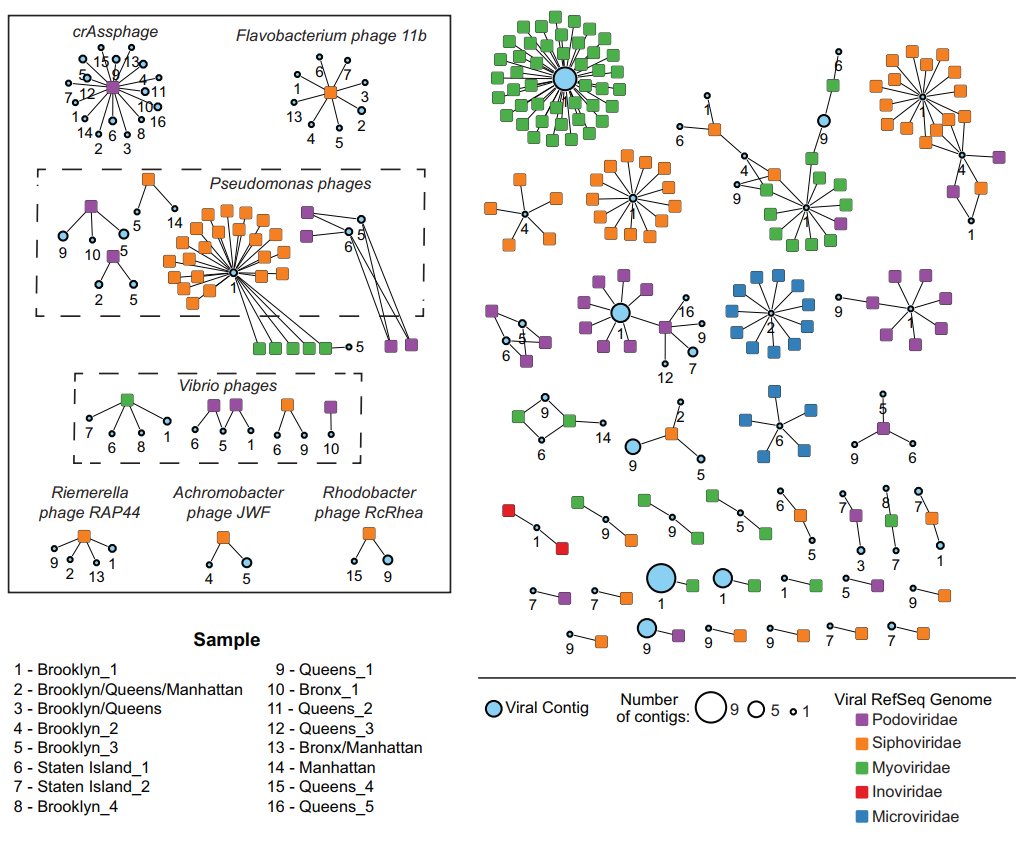
图3:与病毒RefSeq基因组相关的病毒簇
除人体外,污水系统中的病毒还可能来自多种来源。纽约市是混合型污水系统,其中径流水,雨水和垃圾共同进入废水系统。为了识别病毒contigs的潜在环境来源,我们将病毒contigs与综合微生物基因组/病毒(IMG/VR)数据库进行比对,后者包含包括废水在内的多个不同来源的病毒宏基因组数据集。比对结果中匹配的前三个来源是来自人类(1511个contigs)、水生环境(1158个contigs)和废水(823个contigs)(图4a)。样本9来自皇后区,是唯一的类群来源与动物相匹配的样本。只有5个样本与生物反应器数据源匹配,而Manhattan/Bronx的13 号样本与固体废物源匹配的contigs丰度最高。这些环境可以根据它们的源进一步划分为特定的类别。例如,我们鉴定出1,598个contigs来源于人类消化系统(图4b)。将水生环境划分为不同的生态系统后,显示577条contigs属于淡水环境,448条contigs属于海洋环境(图4b)。16个样品中有12个样品的来源与活性污泥相来源匹配,活性污泥是废水处理过程中常见的组成部分。此外,Staten Island的7号样本是唯一与堆肥环境来源相匹配的样本。总的来说,这些数据表明本研究种的病毒contigs来自不同的环境来源。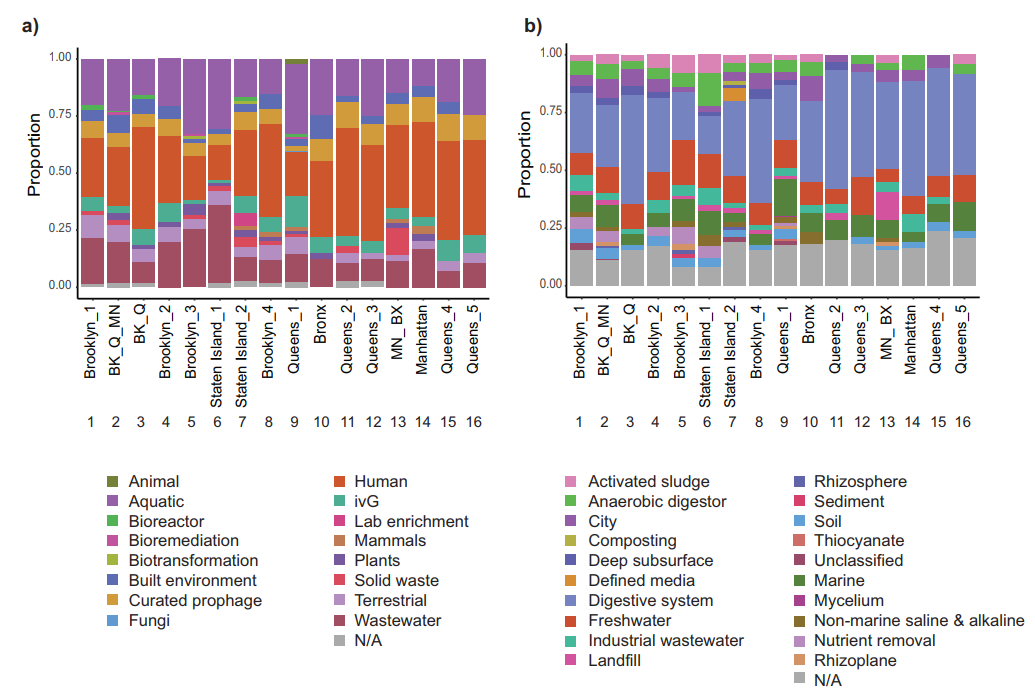
图4:病毒contigs的环境来源
噬菌体含有一组用来感染寄主、复制基因组并产生新后代的基因。除了这些基因,噬菌体基因组还可能携带其他基因,这些基因可以影响其细菌宿主,通过提供代谢或适应优势来帮助整体生存。为了探测该数据集中病毒contigs的功能潜力,我们首先使用UniRef50数据库对预测的ORFs进行注释。样本中的丰度排行靠前的GO注释均与噬菌体有关,包括生物过程,如DNA整合和复制;细胞成分,如病毒衣壳;以及分子功能,如ATP结合和内切酶活性。作者还将UniRef50比对结果映射到MetaCyc酶促反应数据库,以确定污水系统中噬菌体的代谢潜力(图5)。在16个样本中,至少有11个样本都存在三种酶——DNA定向的DNA聚合酶、溶菌酶和核糖核苷二磷酸还原酶。有些酶只对少数样品有特异性。例如,核苷酸二磷酸酶仅在2个样本中存在(Brooklyn_1和Queens_1),而参与碳水化合物代谢的三磷酸尿苷 -葡萄糖-1-磷酸尿苷转运酶,仅在Queens的2个样本中存在(Queens_4和Queens_5)。
图5:病毒contigs中代谢通路基因相对丰度热图
一些噬菌体可以整合到细菌基因组中,它们被称为前噬菌体。前噬菌体可以影响细菌宿主的适应度和毒力。它们可以编码辅助代谢基因,并诱导很大一部分细菌遗传多样性。作者使用前噬菌体搜索工具PHASTER在样本中鉴定了140个前噬菌体相关的contigs。
为了确定在城市环境中噬菌体与宿主的相互作用模式,作者鉴定了CRISPR spacer和direct repeats。每个样本平均鉴定出20,735个spacer和1,686个repeats。Brooklyn/Queens/Manhattan的样本中spacer和repeats序列最多(分别为38,687和2,929)。这可以归因于这三个行政区均含有大量的超越其他样本的污水流。作者成功地为91对噬菌体-寄主配对分配了特定的噬菌体类别(图6a)。在这91对噬菌体-寄主配对中,只有8个噬菌体被鉴定感染它们分类学上指定的宿主。如:链球菌噬菌体315.2与链球菌配对,Geobacillus噬菌体GBSV1与Geobacillus配对。寄主范围最广的噬菌体是Lactococcus phage 1706,它与8种不同的细菌属有关联,这表明该噬菌体可能在城市污水环境中具有广泛性。这些结果表明,在城市污水环境中,噬菌体-宿主的相互作用是广泛的,甚至可以跨越属。
研究团队利用CRISPR spacers可以有效地实现病毒contigs与宿主配对,确定噬菌体-宿主感染模式的潜在网络结构。将上述基于基因的网络分析应用于关联细菌宿主的929个病毒contigs,其中285个VCs可感染102个不同细菌属。作者使用这个二进制相邻矩阵来计算噬菌体-宿主感染网络的嵌套性和模块化。嵌套式网络代表了具有可感染易感宿主的噬菌体层次结构的网络。在一个嵌套式网络中,宿主范围广的噬菌体可以感染所有宿主,而专一噬菌体只能感染一个宿主。在模块化网络中,感染发生在同一亚群的噬菌体和细菌之间,而不是发生在不同亚群之间。这种相互作用模式可能表明独特的噬菌体-宿主感染集群。图示显示感染网络的嵌套性分值较低,同时发现感染网络有着58个优良的模块化化结构(图6 b和c)。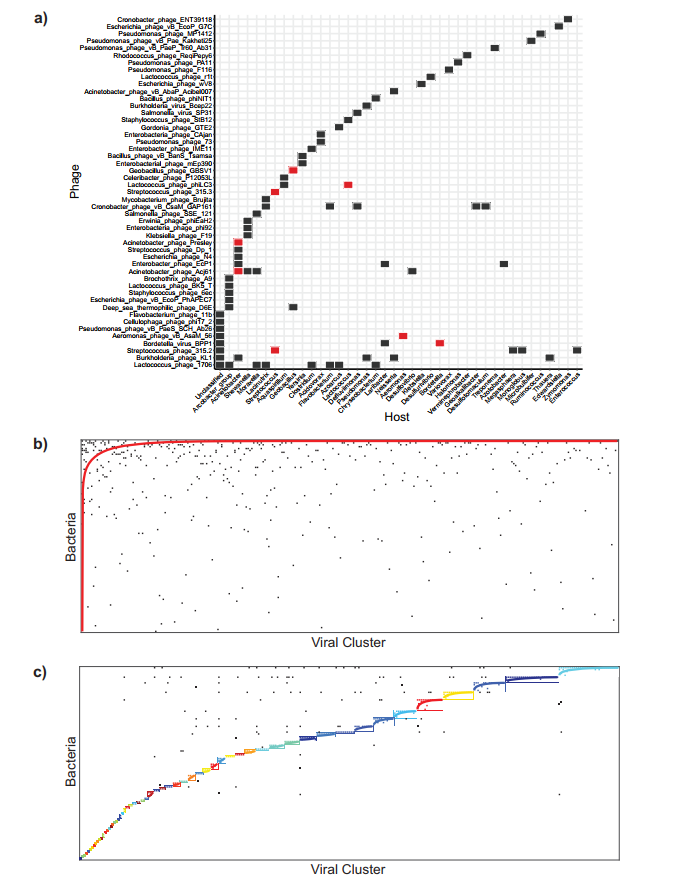
图6:噬菌体-宿主互作关系图
结论:
作者利用2014年11月从14个污水处理厂收集的废水样本的宏基因组数据,对纽约市(NYC)废水中的病毒群落进行了分析。研究发现噬菌体是污水样本中最主要的病毒成分,特定的病毒群落与采样行政区内的当地环境条件有关。绝大多数病毒序列在公共数据库中没有检测到同源匹配序列,平均获得1700个独特的病毒簇(假定属)。研究通过modular-nested模式构建了感染网络,网络表明噬菌体具备宿主特异性。研究还在病毒群落中鉴定了参与碳和硫循环的基因,表明病毒在废水环境中的循环途径和基因功能的重要性。此外,研究鉴定了一系列病毒噬菌体的基因以及一个新病毒噬菌体基因组的近完成图。这些发现有助于了解纽约市废水中噬菌体的丰度和多样性,并进一步研究城市环境中噬菌体-宿主关联的地理模式。
参考文献:
Gulino K, Rahman J, Badri M, Morton J, Bonneau R, Ghedin E. Initial Mapping of the New York City Wastewater Virome. mSystems. 2020 Jun 16;5(3) pii: 5/3/e00876-19. doi: 10.1128/mSystems.00876-19.
文中病毒组研究方法:
- 该研究特色为从宏基因组数据中挖掘病毒组学数据。
- 组装注释采用的软件是VirMAP,该是一个对宏基因组测序数据中的病毒进行检验和分类的综合型分析工具。
- 病毒序列预测采用的软件是VirSorter,软件配套使用pfam数据库。
- 通过blastp及hmmsearch鉴定主衣壳蛋白(MCPs)鉴定相应的噬菌体病毒序列,对候选contig序列进行三个核心基因的检测,包括小衣壳基因、半胱氨酸蛋白酶基因以及DNA包装基因。
- 噬菌体宿主关系预测的方法为使用Crass软件确定CRISPR spacers和direct repeats。







