精确探索功能蛋白质氨基酸序列,这个小软件不可少
丁香园
探索蛋白质发挥功能部分氨基酸的序列规律在生物化学研究中是非常重要的。目前,序列标志(Sequence Logo)是最常用的可视化手段。其产生的结果是一个直方图样式,其中每个栏都用垂直的字母堆叠代替。
堆叠的总高度以位表示,堆叠中每个字母的高度与其在该特定位置的频率成比例。但是,序列标志的不足是假设每个氨基酸发生的概率相同,为 0.05(5%),这显然是不合理的。
而 Icelogo 则可以进行运算以校正用户定义的氨基酸频率,能够帮我们更准确地进行肽段序列分析。

图 1
iceLogo 使用参考组(Reference set)来计算实验组(Experimental set)中每个位置上每个氨基酸发生的几率(p 值)。因此,参考组的选择非常重要,应该反映预期的技术和生物背景。创建的参考集有三种:
第一,静态参考集(static reference set)使用从 UniProt 或 Swiss-Prot 数据库中提取我们所关心物种的特异性序列来计算每个氨基酸的频率。但是,这种一般的蛋白质组参考集可能不会反映预期的生物或技术背景。
第二,我们自己定义的肽序列也可作为参考组。例如,在分析来自磷酸化蛋白质组学实验的数据时,非磷酸化肽就可以构成一个很好的参考组。
第三,可以在 FASTA 格式的蛋白质序列数据库中进行参考集采样,该方法既有优点又有缺点。主要缺点是时间成本,因为在采样时反复迭代,是一个繁琐的计算过程。
其最大的优点是解决了在使用静态氨基酸频率时,假设实验数据集中的氨基酸使用量与整个蛋白质组的氨基酸使用量相同的问题。另一个优点是可以针对预期的技术或生物背景进行特殊定制的参考集。
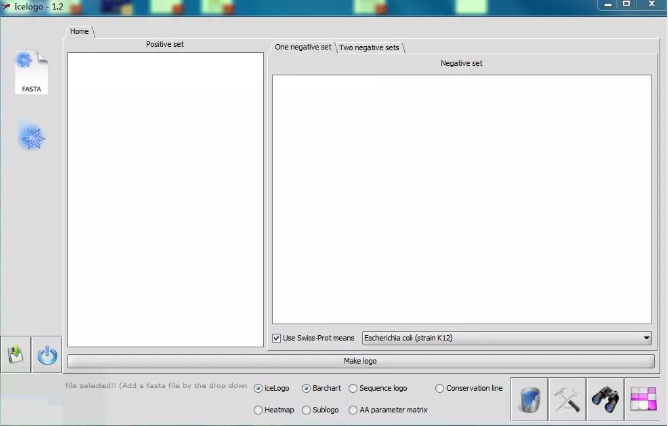
图 2
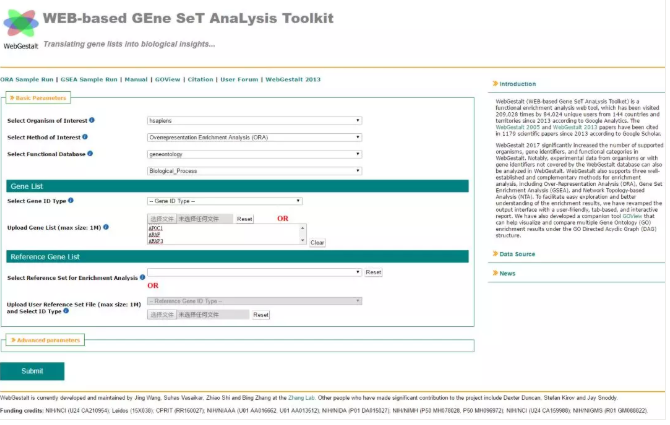
当我们点击时,就是采用静态参考集(图 2)。
正集(Positive set)应该是多序列对齐的格式。感兴趣的区域必须始终位于每一行的相同位置,并且多序列比对中的不同序列必须具有相同长度。
有两种不同的方式来创建静态参考集(Negative set)。可以使用单参考集(One Negative set)或双参考集(Two Negative sets)。
双参考集仅适用于在某些位置应用某种技术偏差的情况,如:在分析序列的第一个位置或最后一个位置时。可以通过使用上方滑块定义第二个参考集开始的位置。可以通过点击使用 Swiss-Prot 平均值复选框和复选框的下拉框中的物种来作为蛋白质组背景。
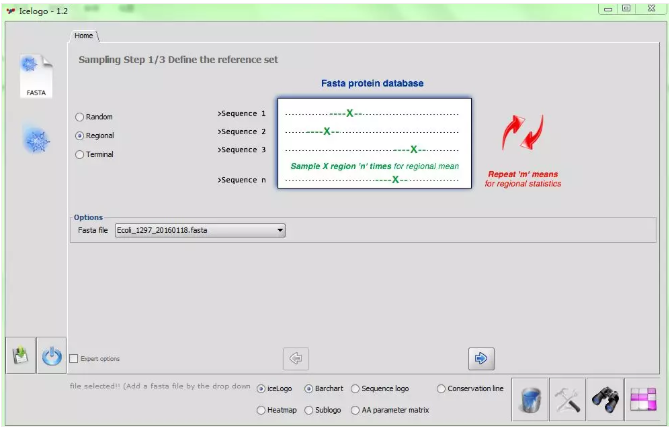
图 3
当我们点击时,就是采用 FASTA 格式参考集(图 3)。
这里需要进行三步设置:
01
设置参考集
有三种参考集的设置方法:
1. 随机抽样方法(Random):利用随机抽样方法计算在 FASTA 中随机遇到某个氨基酸的概率。该算法从 FASTA 中随机读取 n 个蛋白质序列。在每个蛋白质序列中,随机选择一个氨基酸并加入到氨基酸计数器中。当向该计数器中加入 n 个氨基酸时,计算每个氨基酸的 FreqAA。将这个过程再次迭代 i 次来估算 μRandomAA 和 σStandardAA 的标准差。
2. 终端抽样方法(Terminal):计算在 FASTA 中与蛋白质末端相距一定距离处某个氨基酸的概率。该算法从 FASTA 随机读取 n 个蛋白质序列。在每个蛋白质序列中,取长度等于实验组中的氨基酸数目(l)的末端肽(N- 或 C- 端)。将氨基酸加入到氨基酸计数器中。当将 n 个末端肽的氨基酸加入到这些计数器中时,计算每个氨基酸的 FreqAAl。并将这个过程再迭代 i 次,估算 μTerminalAAl 和σTerminalAAl 的标准差。
3. 区域抽样方法(Regional):计算在锚定实验位置周围区域遇到某种氨基酸的概率。该算法首先分析实验组中锚定位置处的氨基酸频率 FreqAAanchor。
例如,锚定的磷酸化位点的实验序列具有 FreqAASer = 70%,FreqAAThr = 30%。算法从 FASTA 中随机读取 n 个蛋白质序列。在每个蛋白质序列中,锚定位点附近区域肽的长度(l)等于实验序列中的氨基酸数。
在本例中,0.70 x n 个区域肽具有 Ser 锚点,0.30 x n 个区域肽具有 Thr 锚点。然后将氨基酸加入到锚定位点周围的氨基酸计数器中。当这些计数器中加入了 n 个区域肽及其氨基酸时,计算每个氨基酸的 FreqAAl。并将这个过程再迭代 i 次,估算 μRegionalAAl 和 σRegionalAAl 的标准差。

图 4
02
输入标题
输入一个对齐的序列集,通过对 FASTA 数据库抽样来创建参考集,并进行测试。该序列通常是实验的结果(蛋白水解切割位点,磷酸化等)。(这里以我随机输入的 25 条序列作为实验集为例,图 4)

图 5
03
参数概述
该步骤不需要进一步输入,而是呈现通过前一步骤收集的我们输入的全部信息(图 5)。
如果没有异议,则可以开始抽样分析,点击「start」按钮。采样分析需要最多几分钟时间。
最后,就得到分析结果了。
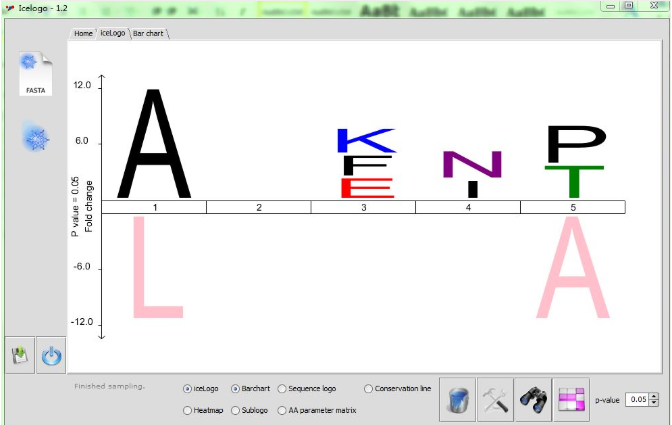
图 6
1. iceLogo 图
iceLogo 可以像序列标志一样全面地可视化研究序列。氨基酸的颜色可以在颜色面板中改变。如果氨基酸发生显著调节,或者如果该特定氨基酸没有发生在阳性或参考集中,则氨基酸将变成粉红色。(图 6)
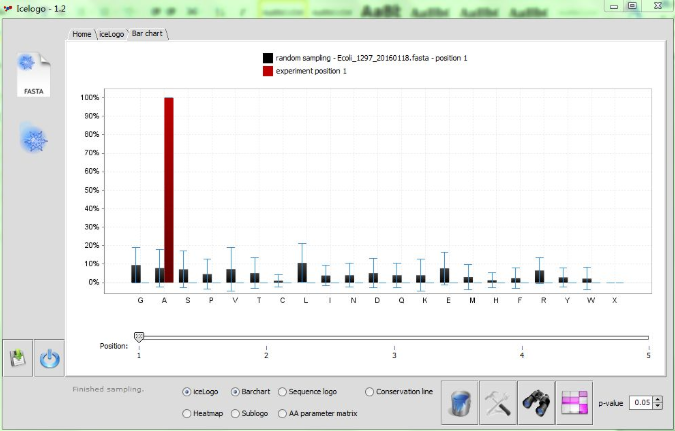
图 7
2. 条形图(Bar chart)
条形图显示所有氨基酸的绝对频率。条形图显示的位置是固定的,因此我们可以通过移动条形图底部的滑块来控制位置。黑条显示从参考集计算的平均值,蓝色误差条显示概率边界(由右下角的 p 值定义)。当使用小的实验数据集时,得到大的标准偏差(如图 7 所示)。 如果实验数据集增长,则误差条缩小。
这样,完整的数据分析与软件功能我们就都非常清楚了,希望对大家的科研有所帮助!