如何分析和展示 RNAseq 基因表达数据中基因的相关性?
优雅 R
介绍
TCGA 是癌症基因组分析中相当流行的数据库,针对里面数据的挖掘结果、软件工具发表了许多 CNS 文章,不过现在已经被整合进 GDC 数据平台了。虽然现在测序技术发展的很快,单个样本测序成本比较之前而言低了很多,但是对于单个课题组研究而言,对大量样本的测序成本还是有些难以承受。
TCGA 的数据集提供了一个很好的平台,我们既可以分析它衍生新的课题,也可以通过它为自己分析的结果佐证。今天的分析用的就是 TCGA 肺腺癌的数据集(TCGA-LUAD),可以点击这里进入 UCSC 的数据集资源库下载。
RNAseq 的结果中包含了数万个基因的表达值,而我们往往感兴趣的只是少数。
基于一些先验知识,我们可能想要查看某些基因之间的相关性如何,以辅助构想这些基因之间的关系模式是怎样的。一种非常直观的办法是对基因两两建立回归模型(线性回归或者广义线性回归)。这样需要画的图和构建的模型根据你想要查看基因数的变化会有很多变化,虽然可以通过循环之类的方式实现,但我并不推荐。懒人表示喜欢简单易懂的,有一种非常简约的办法:构造基因表达的相关系数矩阵,然后展示它。
R 实现
下面看怎么用 corrgram 包实现:
首先构建两个用来读写 tsv 文件(table 键分隔的文件,TCGA 数据集以这种格式存储)的函数。
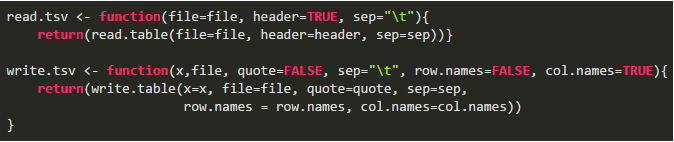
非常简单,就是封装了一下 R 本身自带的 write.table 与 read.table 函数。因为我并没有看到 R 自带 tsv 文件处理的函数,所以封装了这两个函数,用起来更方便。
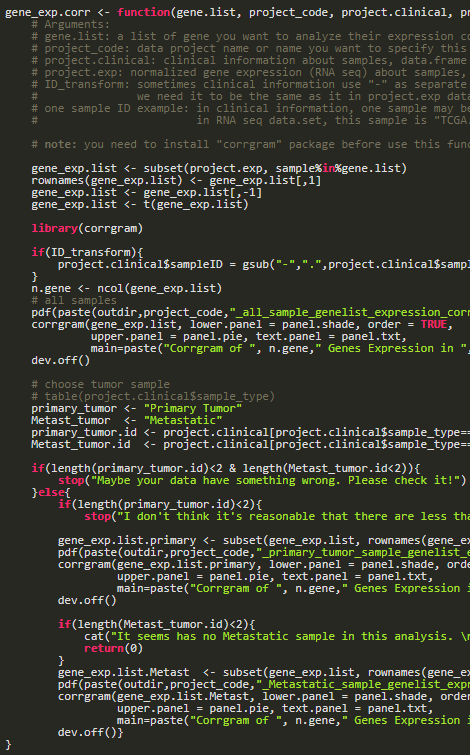
构建一个函数来实现展示基因表达量相关性的功能,它主要完成 3 件事情,根据输入参数提取出进行分析的数据集,将这个数据集作为参数传入 corrgram 函数,然后将生成的图形输出。corrgram() 函数自动会对传入的数据集变量进行相关分析,然后生成图形,所以我们没必要在此之前用 cor 函数处理。
需要传入函数的参数有 6 个,必要的有 5 个。所有参数的含义我已经用英文进行了注释说明(我的 Rstudio 用不了中文)。
简单解释一下,gene.list 是想要查看的基因集(最后只会找出 RNAseq 基因 ID 中能找到的);project_code 是项目代码,比如 TCGA-LUAD,这里也可以当 job id 设置,用以区分;project.clinical 与 project.exp 就是临床信息数据集和 RNA 表达数据集(如果引用非 TCGA 数据集时变量名对不上,自己改下哈);outdir 设置输出文件目录;ID_transform 用来控制样本 ID 的转换,TCGA 临床数据用的是-分隔样本 ID,而 RNAseq 结果中用的是. 分隔,所以需要转换。如果参考使用下面函数时有什么问题,争取自己动手改改,也可以文章下方留言。
因为 RNAseq 数据中包含的病人类型不一,所以在分析所有样本后,我增加提取癌症病人的代码,主要是原位瘤和转移瘤。前者在我见过的 TCGA 数据集肯定有,后面则不一定,所以用 if 语句控制了下分析流程。
下面拿数据实测一下。
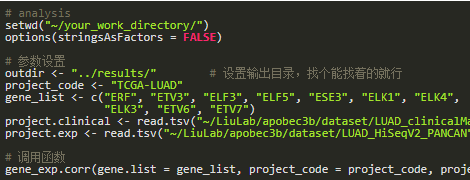
结果以 pdf 格式保存,毕竟矢量图质量好。
还会返回:
![]()
因为这个数据集没有转移瘤病人。
看看输出的图形结果吧,这里只放一张原位癌病人的图当做 demo。

关于图形的输出效果可以参考 corrgram 包参数(help 一下)设定,《R 实战》书中有它的介绍。这里设定的是下三角用阴影图,上三角用饼图,两种结果的解释是一致的。

左下角的方块图和右上角的饼图显示的结果完全相同(展示的是变量(基因)相关矩阵):
蓝色和从左下指向右上的斜杠表示两个变量正相关。反过来,红色和从左上指向右下的斜杠表示呈现负相关。色彩越深,饱和度越高,说明变量相关性越大。
右上角的饼图展示同样信息。颜色功能同上,相关性大小是由被填充的饼图块的大小来展示。顺时针填充为正相关,逆时针填充为负相关。
corrgram 包的 corrgram 函数设置了 order = TRUE,相关矩阵会使用主成分分析方法对变量重排,有点聚类的效果,展示了变量的相关关系模式。