HNADOCK - 一个研究 RNA 与 DNA/RNA 互作的平台
i 生信
核酸(RNA/DNA)之间的相互作用在许多基本的细胞活动中起着重要作用,例如转录调控,RNA 加工和蛋白质合成。因此,确定 RNA/DNA 之间的复杂结构对于理解相关核酸相互作用的分子机制至关重要。
因为结构往往决定了分子的功能,这种核酸-核酸相互作用的结构建模和预测对于理解原子级相关生物过程的分子机制,进而开发针对相互作用的干预性治疗药物或措施也是至关重要的。因为采用实验技术来探究 RNA/DNA 之间相互作用通常是复杂困难且高成本的,因此从核酸序列计算并预测核酸复合体结构并进行分子模拟对接,是目前的有效方式。
在这里,我将介绍一款用于核酸对接的用户友好型在线平台 HNADOCK,它由华中科技大学的 Yi Xiao 和 Sheng-You Huang 团队开发。HNADOCK 可以用于建模两个核酸分子(RNA/DNA)之间的 3D 复合体结构。HNADOCK 的速度很快,通常可在 10 分钟内完成工作。
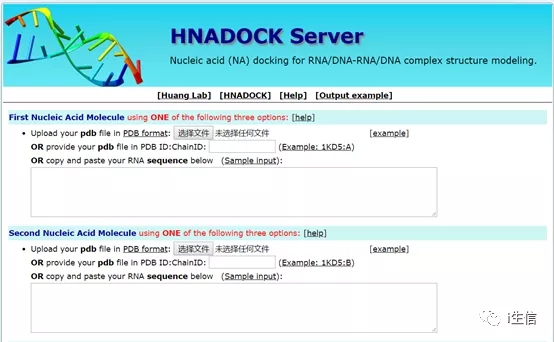
HNADOCK 在线平台主页
图片来源:HNADOCK 官网
HNADOCK 服务器的工作流程包括四个阶段:
数据输入;
同源 RNA 搜索;
结构建模;
基于 FFT 的全局对接
1. 数据输入
要求用户提供要对接的两个分子的三维(3D)结构,其中 RNA 接受序列和 PDB 结构文件(或 PDB 登录号如 1KD5:A)的输入,而 DNA 仅支持 PDB 结构文件(或 PDB 登录号如 1KD5:A)的输入。用户还可以选择提供结合位点信息(HNADOCK 可以自动整合结合位点信息),以及选择是否优化前十个模型。指定结合位点残基可以使预测的模型准确性更高。用户可以提供两种类型的绑定站点信息。
(i)结合位点残留在受体或配体上:在文本框中如下指定结合位点残基
15:A,23 - 26:A,18:B
代表链 A 的残基 15、23 - 26 和链 B 的 18。注意行中的残基必须用逗号分隔。
(ii)相互作用残基之间的距离限制:用户可以在文本框中提供此类信息,例如
15:A 26:B 8,25 - 28:A 36:B 6
表示其中受体上的链 A 的残基 15 和配体上的链 B 的残基 26 之间的距离在 8A 之内;受体上链 A 的残基 25 - 28 与配体链 B 的残基 36 之间的距离将在 6 A 之内。
然后,服务器将检查核酸分子的输入类型。如果输入是结构,则工作流将自动进入最后的对接阶段。如果输入是 RNA 序列,则将通过比较建模或从头计算 RNA 结构预测法来构建其结构。
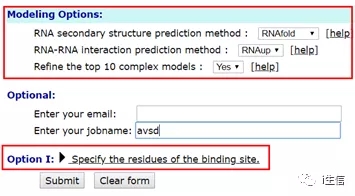
图片来源:HNADOCK 官网
2. 同源 RNA 搜索
首先使用 ViennaRNA 程序包的 RNAfold 程序生成上传 RNA 的二级结构,接着使用基于二级结构的 RNA 比对方法 RADAR / RSmatch 对来自 PDB 的 RNA 结构数据库进行同源性搜索,以找到可能的 RNA 序列同源模板,然后用具有评分功能的内置软件 DITScoreRR 对 RNA-RNA 相互作用进行评估。
图片来源:HNADOCK 官网
3. 结构建模
如果顶部 hit 的比对得分大于 0,则该 hit 将用作 RNA 的模板,然后使用比较 RNA 建模程序 ModeRNA 构建相应的 3D 模型。
由于 PDB 数据库中 RNA/DNA 结构的数量有限,有时难以找到合适的 RNA/DNA 结构模板(即比对得分小于 0),因此 HNADOCK 还可以利用从头算法 3dRNA 以及许多第三方程序来进行快速的 RNA 的结构建模,其中嵌套的 RNAup 和 RactIP 可以用于 RNA 相互作用预测,RSmatch 可以用于同源性搜索,ModeRNA 用于单个 RNA 的比较建模,AMBER 用于结构优化。
4. 基于 FFT 的全局对接
利用用户上传的或 HNADOCK 服务器建模的 3D 核酸结构,HNADOCK 的工作流程进入最后阶段,即核酸对接。对接过程是完全自动化的,如果提供了电子邮件地址,则通过邮件通知和交互式网页将结果呈现给用户。
首先,基于分层 FFT 的全局对接程序 HDOCKlite 用于采集一种核酸相对于另一种核酸的推定结合模式。采用具有 RNA-RNA 相互作用评分功能的 DITScoreRR 软件来评估和排序生成的结合模式。在 RNA-RNA 相互作用预测中,HNADOCK 会将 RNA 间碱基配对信息考虑在内,可显着提高对接准确性。
如果用户在提交任务时提供了此类信息,则对接过程还将包含结合位点信息,即施加限制以确保相应的核苷酸位于同一界面上或在一定距离之内。用户还可以选择可以通过 MD 仿真来细化预测的前 10 个模型,但任务将花费更长的时间才能完成。
5. 输出结果
任务完成后,网页将自动显示对接结果以供可视化和下载。对接结果包括两种类型的文件:单个 PDB 文件和对接的复合体模型。
(i)用户上传或由服务器根据用户提供的 FASTA 序列构建的受体和配体 RNA/DNA 的 PDB 文件。
(ii)服务器会为每个任务预先生成前 100 个结合模型。用户可以单独下载前 20 个结合模型中的任何一个,也可以选择将所有前 10 个预测模型或前 100 个预测模型打包下载。
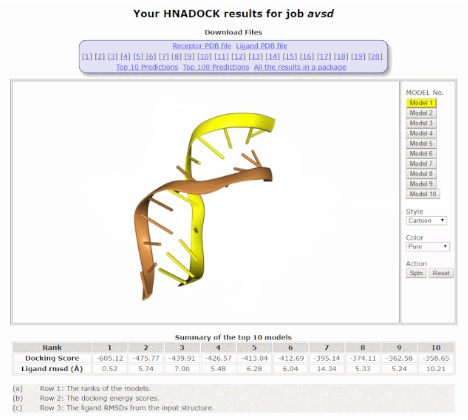
HNADOCK 服务器结果页面。
页面顶部是任务名称及 ID,以及要下载的文件。
右侧的可选按钮可以控制 NGL 可视化视窗的结合模型。前 10 个模型的对接得分在底部显示。
由于通常认为前十个结合模型是大分子对接中最重要的模型,因此结果页面还使用 NGL 查看器提供了前十个模型的交互式可视化。用户可以选择以不同的颜色、形式或样式查看排名前 10 位的模型中的一个,或多个一起查看。
结果页面还提供了前 10 个复合体模型的排名和对接分数,其中分数基于对 RNA-RNA 相互作用的评分的 DITScoreRR。但应注意的是,此处的对接分数并不能反映真实的结合亲和力,而是复合体模型的相对排名,因为 DITScoreRR 并未使用实验性结合数据进行校准。
HNADOCK 官网:http://huanglab.phys.hust.edu.cn/hnadock/
参考文献:
Jiahua He, Jun Wang, Huanyu Tao, Yi Xiao,Sheng-You Huang, HNADOCK: a nucleic acid docking server for modelingRNA/DNA–RNA/DNA 3D complex structures, NucleicAcids Research (IF = 11), Volume 47, Issue W1, 02 July 2019, Pages W35–W42,https://doi.org/ 10.1093 /nar/gkz412

![十二烷基二甲基(3-磺丙基)氢氧化铵内盐 [用于生化研究],14933-08-5,≥98%,阿拉丁](https://img1.dxycdn.com/p/s14/2024/0619/475/6370229598169633081.jpg!wh200)





