基因克隆的流程
丁香园
研究某个 gene 的功能通常要做相关的功能实验,常用的手段有 gain-of-function,涉及 gene 的过表达,在下文中即将讲述;还有 loss-of-function, 通常涉及 gene 的 knock-out 或 konck-down,暂且不提。
今天准备跟大家分享一下如何成功地用传统酶切酶连的方法克隆某 gene 的经验,虽然现在同源重组的方法已经越来越广泛地应用到科研中,属于 technology based innovation,给实验带来极大便利。
但是传统的克隆方法还是占主要地位,尤其是新兴的方法如果要做 gain-of-function 仍然逃避不了克隆 gene 片段这个步骤,打好相关的基础,掌握其中原理才能在各种 tools 出现时及时掌握并灵活应用。
接下来讲述基因克隆的流程
1 首先获得某基因的序列
在 pubmed 主页下拉框选定 Gene 或者 Nucleotide,然后在输入框输入基因名称如 Znf516 点击 search 后,进入(如图 1)。
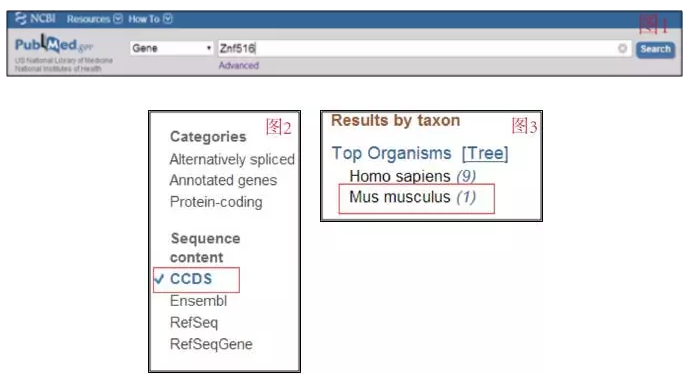
在左边分类 Sequence content 里选择 CCDS(如图 2),在右侧选择种属 Mus musculus(如图 3), 这样就只会显示此基因的 CDS 结果。而页面加载出来后会有很多 Znf516 相关的信息包括基因名,全称,在 chromosome 上的位置及其相邻的基因有哪些,目前已知的此基因相关文献报道,可以忽略这些信息(当然也可以留意一下自己比较关注的那部分内容),在右侧栏目直接点击 CCDS,这样就进入了一个页面,在页面最下方是这个基因的 CDS 的核苷酸序列及蛋白翻译的序列。
可以将此序列 copy 到 snapgene 软件里即完成步骤一。
2 用 Snapgene 设计引物
注意:要看清楚载体上 promoter 的方向,将 CDS 正向插入到 promoter 后面的 MCS 中间。想要 snapgene 软件?请在对话框回复基因。
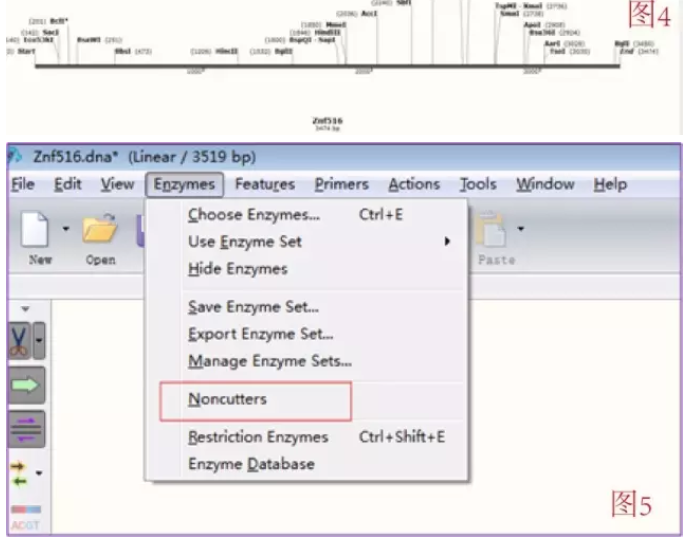
比如说 MCS 上的酶切位点是 ABCDE 五个酶切位点,那么设计引物是必须将 A 酶切位点加在 CDS 上游,B 加到 CDS 下游,不可颠倒。并且,最好保证选择的两个酶切位点在载体上不要相邻,如若必须如此则在切载体时先切割对保护保护碱基要求高的位点(请自行百度 NEB 酶切位点保护碱基,查保护碱基个数及切割效率),再切另一个位点。这样可以提高酶切效率。
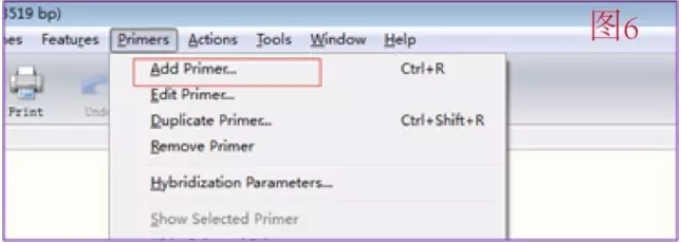
这样就可以开始设计引物:用鼠标拖选 Znf516 前 28-25 bp(可以通过页面最下面一排的标题栏选择 Sequence,这样可以看到一个个的核苷酸序列,方便拖选,而且在拖选的时候会显示多少 bp 的序列被选中,GC 比以及 Tm 值,很方便), 一般 Tm 值为 50-60C 都是可以接受的。点击 Add primer(如图 6)。

这样就跳出提示框,是需要选中序列的 Top strand(正向序列) 还是 bottom strand(反向互补序列),对于正向 / 上游引物,选择正向序列(若是反向 / 下游引物,选择反向互补序列)。
比如我要设计 Znf516 正向引物,并加 XbaI 的酶切位点,那么如图 7:选择 Insertions 中第二行 site 下拉框中的 XbaI,点击 Insert。即将酶切位点(红色大写字母的 6 位碱基)加到了引物上游(如图 8),一般在酶切位点旁边加保护碱基(NEB 主页有优化的保护碱基序列,初学者可以注意一下,克隆做熟练了发现其实不必如此苛刻。根据 GC 比,如果 GC 比正常 55%,或偏低,加两个 g, 如果高则加若干 t)。
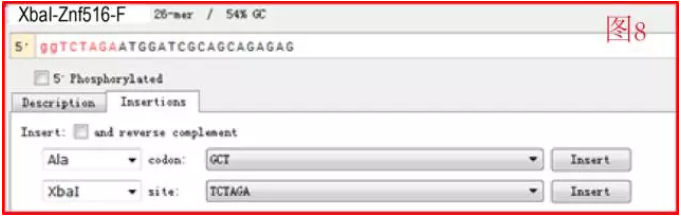
引物的命名应该规范一些这样之后在实验室管理及整理时比较方便,一般如下:酶切位点 - 种属缩写 - 基因名 - F/R
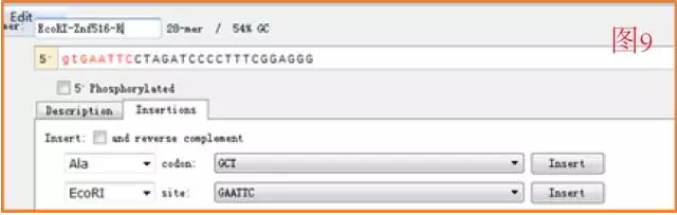
反向引物设计如图 9。
这样引物这一步骤完成了,送到生工合成即可。我们 Lab 现在统一用 synbio,新兴「国产」公司,价格便宜且态度优良,更可贵的是支持晚上 11:00 下单,想必大多数生物汪都会因此喜欢上它。
3 寻找合适的模板
找到高表达这个 gene 的 tissue, 在 gene 表达丰度高时后期实验更易成功,这个可 refer 各种 paper, 或者 BioGPS website,非常 powerful。
使用方式大概是:打开网站,输入 GeneX,点选 species, 之后就出来各种细胞系或者组织表达此 geneX 相对量,找到表达 X 高的组织用其的 cDNA 当模板来做 PCR。
如果遇见要 P 组织特异性的 gene 的 variant,那么提高此组织 cDNA 的量或者两轮 PCR(第一轮 P 万后用作模板再 P 一次)都是可行的。

4 获得目标片段
例如从 forebrain cortex 组织的模板上 P 这个 gene, 我们使用的是 KOD 酶(Takara,日产,酶中「墨渊」)。改变很多 PCR 条件都无法 P 出这个 gene(之所以今天举这个栗子,原因就在于此),后分析发现这个 Znf516 平均 GC 含量 61%,局部的 GC 含量高导致 P 出片段不对。此时师兄光环笼罩,指点小僧道: 加 DMSO 有助于 P 高 GC 的基因,遂悟。我的经验做法是 100ul 体系加 0.8uL。
得到目标序列后,切胶回收(紫外照射对身体尤其眼睛不好,给 DNA 胶插上几个枪头确定坐标,在照胶时注意目的条带的位置,之后再关掉紫外去切胶避免手和胳膊及眼睛被紫外毒害,可谓是「穷苦」实验室首选妙法)。

5 酶切连接转化
酶切回收片段及载体后,将载体:DNA 片段 = 1:8 加到连接体系,16 度用 T4 酶连接 15 min,或更久,转化到感受态即可。
6 鉴定:挑克隆摇菌
如果赶时间的话可以边挑菌边鉴定。用 Taq Premix 做鉴定即可。鉴定完送测菌液。之后找公司索要正确序列对应的质粒,至此就得到了您想要的克隆。
最后可能会遇见的问题,在实验过程中如果遇见要构建的蛋白没有很好用的抗体,或者做体外 co-IP 等相关实验,需要构建一个融合蛋白,方便之后用 tag 检测:可以利用软件将 CDS 翻译为蛋白,必要时添加碱基,使目标 CDS 与 tag CDS in frame(中间间隔的 linker 是 3 的倍数,因为编码是以 3 个碱基一组的);如果 tag 在下游,要去掉基因上的终止密码子。