小鼠模型,tau蛋白缠结和阿尔茨海默病的神经变性
最常见的神经退行性疾病有阿尔茨海默病、帕金森病、运动神经元病等。这些疾病的行为症状可能各不相同,但都以神经元的进行性损伤和死亡为特征。成年人的神经元不容易被替换,因此神经元损伤会导致患者的身体逐渐衰弱,因为运动或精神功能逐渐恶化。

目前还没有治愈任何神经退行性疾病的方法,尽管我们知道有一些共同的主题——蛋白质错误折叠、线粒体功能障碍、微管损伤——这些是如何导致疾病的仍然是未知的。
对这个问题的研究是贯穿今年神经科学学会第46届年会的一个主题,一系列研讨会不仅展示了我们所学到的知识,还展示了使新见解成为可能的工具。阿尔茨海默病是最常见的痴呆症,是一个特别突出的话题。
小鼠模型
研究神经退行性疾病的核心工具是概括其进展的小鼠模型。但这并不容易实现。
例如,直到最近,大多数阿尔茨海默病的小鼠模型都是基于病理学中涉及的人类基因的过度表达,尤其是淀粉样前体蛋白(APP)。APP被分割成几个片段;1、Aβ(特别是Aβ42形式)阿尔茨海默病的斑块特征形式,和被认为是疾病进展的重要中介。
但出现症状早在这些模型,方便研究人员,但不是人类疾病的特征,从他们在其他方面也有所不同,并受到过度的批评的水平未知人类疾病可能引起应激反应,尤其是不仅Aβ,但是其他应用程序片段(不清楚)的生物功能也正在增加。
为了解决这个问题,Takaomi Saido和他的同事创建了一个新的模型,他们在2014年发表的一篇论文中描述了这个模型——“APP敲入”,在这个模型中,他们没有过度表达一个新基因,而是将与阿尔茨海默病相关的突变引入内源性APP基因。这赋予典型特征的疾病,包括与年龄有关的记忆障碍、神经炎症,和Aβ积累/聚合,导致斑块沉积,在生理水平的基因表达。
这些小鼠被免费分发给其他研究阿尔茨海默病的人,两年之后(症状在6到18个月的时间内出现),在今年的神经科学学会会议上,有整整一场会议专门针对APP敲入小鼠的后续调查。
Ilya Bezprozvanny (UT Southwestern)谈到了他最近在APP敲入小鼠研究中所做的工作,即钙代谢异常在阿尔茨海默病中突触脊柱缺失中的作用。从以前的工作中有证据表明,阿尔茨海默病中的记忆衰退是由海马神经元中的突触刺——特别是一种被称为蘑菇刺的种类——缺失引起的。蘑菇脊柱稳定性依赖于突触钙离子/钙调蛋白激酶的活性二世(CamKII),因此适当的Ca2 +的水平,进而取决于Ca2 +进入神经细胞通过神经元门店Ca2 +条目(nSOC)频道,这是由基质相互作用分子2 (STIM2)。
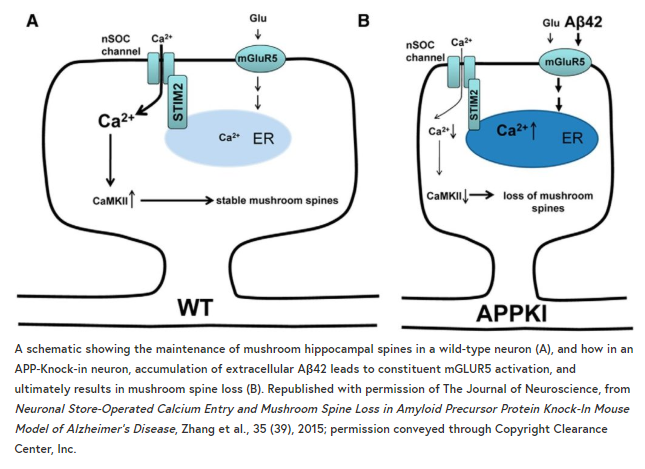
这张照片,Bezprozvanny和他的同事们已经添加了一个影响Aβ42 mGluR5受体的培养从应用敲入小鼠神经元。Aβ42积累导致over-activation mGluR5受体,进而在内质网Ca2 +水平升高,从而STIM2下调。这进而导致通过nSOC流入的减少和CamKII激活的减少,最终导致海马神经元蘑菇棘的丢失。
支持这一机制的是,他们发现抑制mGluR5或STIM2的过度表达,可以挽救APP敲入神经元中蘑菇棘的丢失。随着报道称STIM2在散发性阿尔茨海默病中下调,这项工作为“神经退行性变的钙假说”提供了支持,并提示nSOC可能为阿尔茨海默病提供一个治疗靶点。
限制和组合
由于很难确定任何特定的分子机制在阿尔茨海默病痴呆发展中的作用,来自人类大脑样本和不同的小鼠模型的支持证据对任何特定的机制都是重要的。
Amantha Thathiah(匹兹堡神经退行性病变研究所)在2015年的一次国际合作中,她和她的同事收集了另一个潜在的治疗靶点GPR3。在他们的研究中,两个应用程序敲入小鼠使用另外两个小鼠模型表明GPR3表达水平与Aβ水平在所有的四个模型,与删除GPR3导致血小板减少和减轻认知赤字负担。
他们还报告说,GPR3在阿尔茨海默病患者死后的脑组织中升高,为阿尔茨海默病病理学中的作用提供了进一步的支持。
APP敲除小鼠的缺陷之一是它们的轻度、后期发展的表型,这使得识别行为缺陷变得困难——因此需要另外两个小鼠模型来检查这种与GPR3表达相关的表型。
APP连锁反应的另一个不足之处(也适用于过表达模型)是它们未能显示阿尔茨海默病或神经元细胞死亡特征的神经原纤维tau缠结。
这些可能在一定程度上反映了使用鼠标而不是人类系统的局限性,并试图探索这种可能性,巴特•德•斯特鲁VIB(/鲁汶KU)将人类诱导多能干细胞植入新生程序敲入小鼠大脑看到人类神经元的影响在这些小鼠阿尔茨海默病病理模型。
这些神经元也未能开发阿尔茨海默病的神经纤维τ缠结特征,但它们确实包含Aβ斑块(虽然在这个阶段,目前尚不清楚这些斑块由生成的鼠标或捐赠的人类细胞,在最初的触发效果),他们经历了大规模坏死的细胞死亡过程。
这表明,人类的神经元可能特别容易受到阿尔茨海默氏症病理的影响,而这些嵌合体提供了一种方法,让我们开始思考为什么会这样。
τ缠结
为了解决缺少缠结的问题,Takaomi Saido和Takashi Saito在会议上谈到了这个问题,他们开发了一个包含所有六种形式的人类tau蛋白的老鼠模型(在老鼠中只有三种形式)。他们再次将他们的老鼠提供给任何有兴趣在他们的研究中使用它们的人。
在这些小鼠中,tau蛋白定位正常,小鼠健康。然而,当它们与应用程序交叉敲入小鼠,τ是过度磷酸化和神经元死亡是明显的,从本质上讲,“人性化”τ导致一个强调疾病的表型(支持的想法Aβτ的上游)。然而,奇怪的是,这里仍然没有缠结。齐藤推测,这可能是因为tau蛋白的小聚集物实际上具有致病性。
了解tau蛋白在体内是如何聚集的,以及聚集与神经毒性的关系,是一个关键问题——不仅在阿尔茨海默病中如此,在以tau蛋白聚集为特征的多种神经退行性疾病中也是如此,比如额颞叶痴呆。
为了实现这一目标,马萨诸塞州总医院布拉德利·海曼实验室的莎拉·德沃斯提出了一种tau传感器,它将提供一种监测tau聚集的方法。他们使用一种基于FRET的方法,用一个单一的载体包含与CFP融合的tau (tauRD)重复域和与YFP融合的tauRD重复域,以确保两者在海马注射后都转染到单个神经元。
DeVos报告说,正常情况下,FRET2 tau在细胞内仍呈弥散状态,但其附加的好处是,在突变的tauopathy原代神经元中可以观察到自发聚集,在活的突变的tauopathy小鼠大脑中,在颅窗的帮助下也可以观察到自发聚集。这使得我们有可能观察到单个神经元在产生tau蛋白聚集后的一段时间内发生了什么,并进行基因表达表型分析,以帮助确定tau蛋白聚集在活神经元中的一些分子后果。
多样化的原因,多样化的方法
正如会议上坦率承认的那样,没有一个模型可以被期望忠实地复制疾病表型。大多数神经退行性疾病是异质性的,功能丧失需要与功能获得性突变区分开来,而最根本的是,我们还不知道疾病的哪些特征反映了疾病进展,哪些只是相关的。解开神经退行性疾病的机制似乎需要我们所有的工具。