炎症促发阿尔茨海默病又添一力证!NLRP3 炎症小体激活引发 tau 蛋白异常
在 AD 疾病进程机制中伴随着 β 淀粉样蛋白 Aβ 的斑块积累、神经原纤维缠结中 tau 蛋白过磷酸化和神经炎症发生。
Aβ 是 AD 患者脑内老年斑的主要成分,也是在神经退行性疾病中发现的第一个可以激活炎症小体的分子。
脑内 Aβ 的沉积可刺激胶质细胞加剧炎症反应。Aβ 激活 NLRP3 炎症小体,引发下游 IL- 1β 的成熟和释放,可能是 AD 的重要发病机制之一。
2019 年 11 月 20 日,Nature 杂志在线发表了题为《NLRP3 inflammasome activation drives tau pathology》的文章,为炎症小体激活驱动 AD 提供了新的证据。

图片来源:nature
阿尔茨海默病
阿尔茨海默病(Alzheimer's disease,AD)是一种起病隐匿呈进行性加重的神经系统退行性疾病。临床表现以记忆障碍和失语、视空间损害及人格和行为改变等全面性痴呆表现为特征。
其基本概念、治疗药物面对的研发进展和困境我们在前文(阿杜卡弩单抗 Aducanumab(BIIB037))中已经提到,这里不再展开。
炎症小体(inflammasome)
炎症小体是由胞浆内模式识别受体(PRRs)参与组装的多蛋白复合物,属于天然免疫系统的重要组成部分。炎症小体能够识别病原相关分子模式(PAMPs)或宿主来源的危险信号分子(DAMPs),招募和激活促炎症蛋白酶 caspase- 1。
活化的 caspase- 1 切割 IL- 1β 和 IL- 18 的前体,产生相应的成熟炎症因子。炎症小体的活化还可诱导细胞炎症坏死(pyroptosis)。
炎症小体的基础结构
炎症小体以 NOD 样受体(NOD-like receptor,NLR)或肝再生增强因子(augmenter of liver regeneration,ALR)蛋白家族作为受体蛋白(receptor)、凋亡相关微粒蛋白(apoptosis-associated speck-like protein containing CARD)ASC 作为接头蛋白(adaptor)、含半胱氨酸的天冬氨酸蛋白水解酶 caspase 作为效应蛋白(effector)。
其中接头蛋白 ASC 包含两个重要的结构域即 PYD 和 CARD。CARD 结构域发挥桥梁作用,连接受体蛋白和效应蛋白,ASC 的 CARD 结构域可与效应蛋白 CARD 结构域结合形成 CARD:CARD 相互作用。
其 N 端的 PYD 结构域与 NLRP3 的 PYD 结构域结合并相互作用,C 端的 CARD 结构域与效应器 pro-casepase- 1 的 CARD 结构域连接,通过募集 pro-caspase- 1 介导形成 NLRP3 炎症小体复合物。
Caspase- 1 的激活促使 IL- 1β 和 IL- 18 等细胞因子前体裂解、成熟和释放,最终导致了细胞(如神经元、神经胶质细胞等)的炎症坏死。
NLRP3
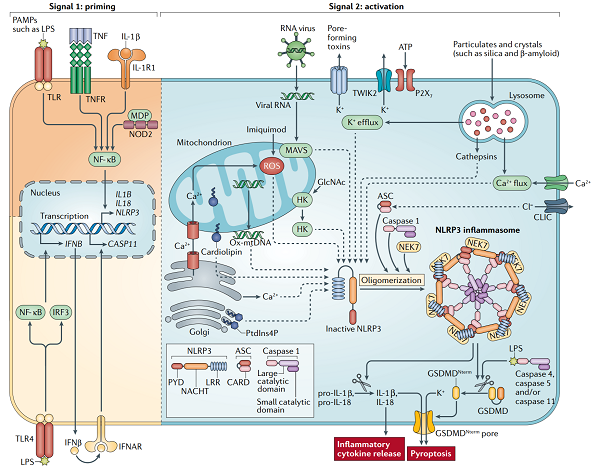
图片来源:Nature
NLRP3 炎症小体具有 NLR 家族成员的经典结构,即核心部分 NLRP3,包括 N 端热蛋白结构域(pyrin domain,PYD),介导同型蛋白相互作用,参与炎症小体激活信号转导过程;
C 端的亮氨酸富集结构域(leucine-rich repeat,LRR),可识别 PAMP 及 DAMP;中央为核苷酸寡聚化结构域(nucleoside triphosphatase domain,NACTH),对核酸连接、蛋白寡聚化及炎症小体复合物的形成发挥着重要作用。
NLRP3 炎症小体作为固有免疫的重要组分在机体免疫反应和疾病发生过程中均发挥重要作用。因此,作为炎症反应的核心,NLRP3 炎症小体可能成为包括 AD 在内的炎症性疾病的治疗提供新的靶点。
研究思路
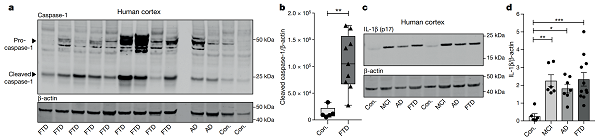
tau 蛋白病、阿尔茨海默病和对照人群脑皮质 caspase- 1 和 IL- 1β 的表达增加
图片来源:nature
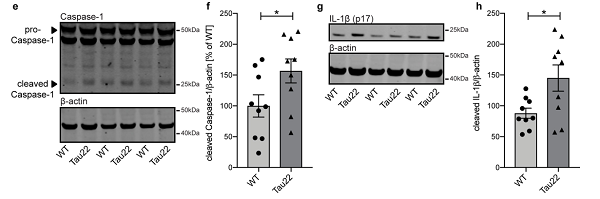
为确认 NLRP3 炎症小体在 tau 蛋白病中的作用,研究人员比较了额颞叶痴呆(FTD)、轻度认知功能障碍(MCL)、阿尔茨海默病(AD)及健康人群的 caspase- 1 和 cleaved caspase- 1 和炎症细胞因子 IL- 1β(炎症小体活化的关键标志)表达水平。
结果表明,与健康人群相比, FTD 患者脑皮质中 caspase- 1 和 cleaved caspase- 1 表达水平显著增高;MCL、AD 和 FTD 人群 IL- 1β 表达水平显著增加,提示在上述 tau 蛋白疾病的发展过程中,存在 NLRP3 炎症小体的激活,表明 NLRP3 炎症小体可能参与 tau 蛋白病的发生。
图片来源:nature
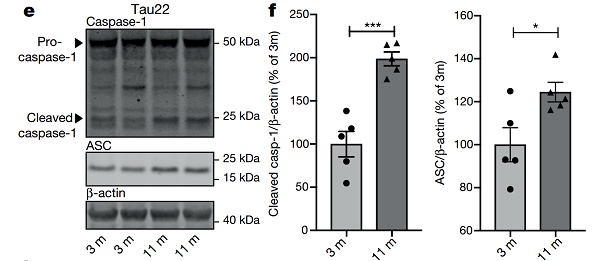
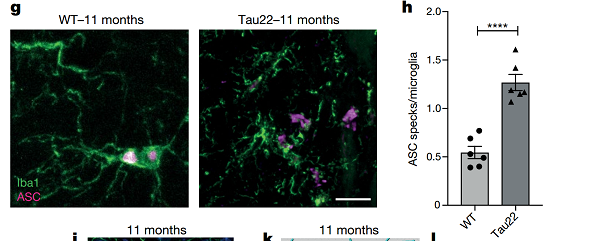
随后,研究人员在表达人类突变 tau 蛋白的 Tau22 小鼠模型中同样观察到 NLRP3 炎症小体活化的表型即 cleaved caspase- 1、IL- 1β 和 ASC 表达水平增加。
注:ASC 是炎症小体中连接胞浆内受体和半胱天冬酶 - 1 的接头蛋白,在炎症小体活化中 ASC 聚集成大分子的二聚体,被称为 ASC 斑点 ASC-speck。
Tau22 小鼠由于 tau 蛋白突变,炎症小体活化,逐渐进展为额颞叶痴呆,提示在 tau 蛋白疾病的发展过程中会诱导炎症小体活化,即 tau 病变和 NLRP3 炎症小体活化密切相关。

图片来源:nature
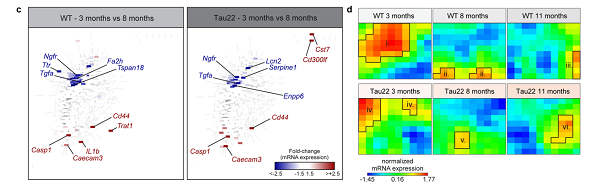
基于上述临床患者人体和实验动物的结果,研究人员深入到基因层面,通过生物信息学分析,比较了不同月龄的 WT、Tau22、Tau22 /Asc−/−、及 Tau22 /Nlrp3−/−小鼠的差异表达基因 DEGs。结果显示脑神经炎症相关基因的表达 Casp1 和 Il1b 基因表达显著上调。
此外,IFNAR1 缺失的小鼠可耐受 Aβ 介导的神经细胞毒性,而两组年龄的 Tau22 小鼠的基因特征表明参与疾病的基因约 73% 与 I 型干扰素有关,提示 NLRP3 炎症小体及其信号通路在 Tau 相关疾病中的关键作用。
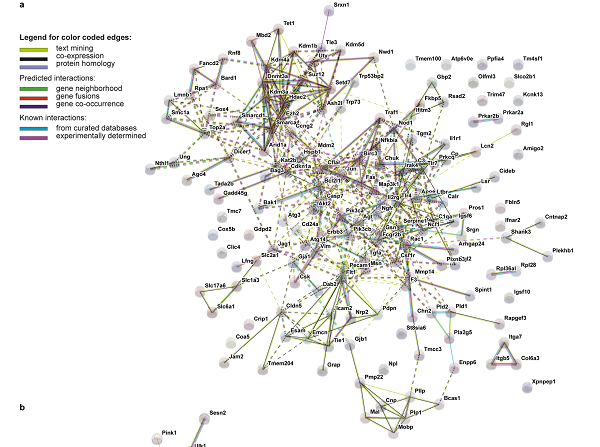
图片来源:nature
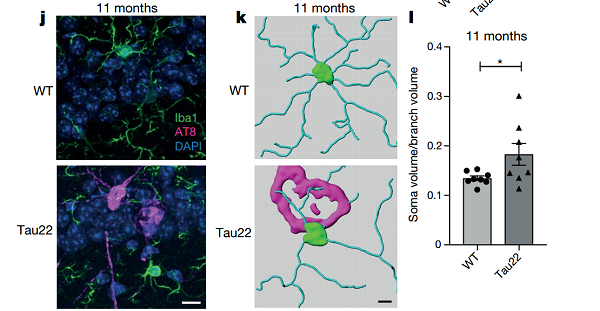
神经胶质细胞差异基因的基因调控网络分析发现,Tau22 小鼠促炎因子相关基因(Jun,Fas 和 Toll 样受体)和染色质重构相关基因(Hdac2)存在相互作用,提示在 Tau 蛋白病早期,神经胶质细胞存在炎症表型和表观遗传层面的调控。同时,Tau22 小鼠胶质细胞存在明显形态学异常。
图片来源:nature
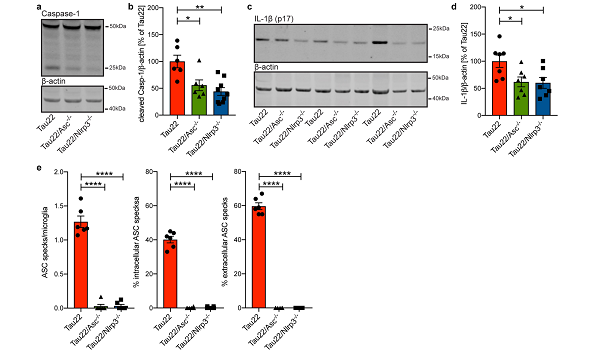
为进一步验证 NLRP3 炎症小体参与 tau 蛋白病的机制,研究人员分别构建了人细胞凋亡相关斑点样蛋白 Pycard 敲除(Tau22 /Asc−/−)和 Cias1 敲除(Tau22 /Nlrp3−/−)小鼠。
实验结果表明 Tau22 /Asc−/−小鼠和 Tau22 /Nlrp3−/−小鼠 cleaved caspase- 1 和 IL- 1β,ASC 表达水平显著减低,表明 ASC 和 NLRP3 介导炎症小体激活。NLRP3 缺失通过抑制炎症反应对 tau 蛋白病具有一定程度的保护作用。

图片来源:nature
Tau22 /Asc−/−和 Tau22 /Nlrp3−/−小鼠的炎症反应显著降低,额颞叶痴呆的病理表型显著缓解,两组保护性敲除的小鼠认知功能降低得到显著改善,上述结果进一步表明突变 tau 蛋白诱导的神经炎症和神经疾病依赖于 NLRP3 炎症小体。

图片来源:nature
Tau 蛋白主要分布在神经元和神经胶质细胞,其磷酸化异常与多种神经疾病相关。正常生理情况,tau 磷酸化修饰有利于微管的稳定。过磷酸化则导致细胞骨架变形和聚集和功能失常。
Tau 蛋白磷酸化是由激酶和磷酸酶的平衡来进行调控。蛋白磷酸酶 2A(PP2A)可使 tau 去磷酸化。糖原合成激酶 GSK- 3β 是催化 tau 蛋白配对螺旋样纤维位点磷酸化的主要激酶。
p25 和 p35 是细胞周期依赖性蛋白激酶 5(CDK5,tau 蛋白过磷酸化的关键酶)的激动剂,而钙 / 钙调素依赖性蛋白激酶 II(CaMKII)可能参与 tau 蛋白的过度磷酸化。
研究人员分析了 Tau22 小鼠、Tau22 /Asc−/−小鼠和 Tau22 /Nlrp3−/−小鼠 NLRP3 炎症小体促进 tau 蛋白过磷酸化的关键激酶和磷酸酶,结果表明,在 ASC 或者 NLRP3 缺失的情况下,诱导 tau 磷酸化的激酶 CaMKII-α 和 GSK- 3β 下调,而抑制 tau 磷酸化的磷酸酶 PP2A 上调,表明 NLRP3 炎症小体通过调控 tau 蛋白过磷酸化的关键激酶和磷酸酶参与 tau 蛋白病的发生。
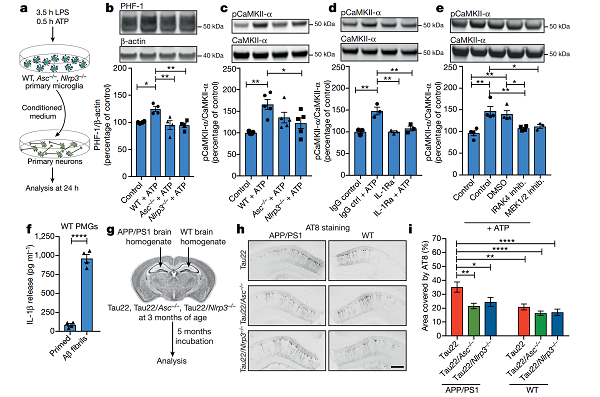
图片来源:nature
众所周知,Aβ 堆积能够促进神经炎症和 tau 病变,前述结果提示 NLRP3 炎症小体促进 tau 病变和炎症发生。
为进一步阐明 Aβ 是通过活化 NLRP3 炎症小体促进 tau 蛋白病的发生,研究人员分离了 LPS 和 ATP 诱导产生 NLRP3 炎症小体的 WT、Asc−/−、Nlrp3−/−小鼠的原代神经胶质细胞,将获得自上述不同组的条件培养基与原代神经元细胞共培养。
来自 Asc−/−小鼠、Nlrp3−/−小鼠的条件培养基未能 Ser396 /Ser404 位点 PHF- 1 的磷酸化水平,而来自 WT 的条件培养基则显著增加神经胶质细胞 PHF- 1 的磷酸化,上调诱导 tau 磷酸化的激酶 CaMKII-α 水平。
此外,抑制神经元 IL- 1 受体或其信号通路下游效应器 IRAK4 和 MEK1 / 2,可抑制 CaMKII-α 介导的 tau 磷酸化,证实了 NLRP3 炎症小体活化和 IL- 1β 在 tau 蛋白中的驱动作用。
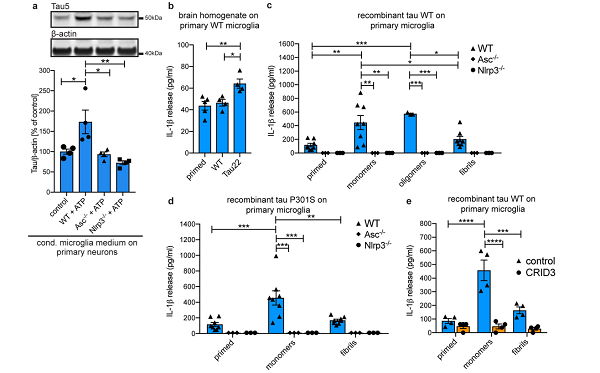

图片来源:nature
此外,应用 LPS 和 ATP 诱导产生 NLRP3 炎症小体的 WT、Asc−/−、Nlrp3−/−小鼠原代神经胶质细胞条件培养基处理原代神经元细胞,WT 神经元细胞 tau 蛋白水平显著增加。
Tau22 小鼠脑组织匀浆获得的 tau 单体和聚体处理神经胶质细胞后,NLRP3 炎症小体显著活化并合成释放 IL- 1β。Asc−/−、Nlrp3−/−小鼠,tau 磷酸化水平和病变特征并不显著,表明 Aβ 诱导 tau 病变发生依赖于 NLRP3 炎症小体。
上述实验结果完整地建立了 AD 中 Aβ、炎症小体活化和 tau 间的关联。
小结和亮点
炎症小体自 2002 年报道后一直是免疫和炎症疾病领域的重点研究方向。
NLRP3 是目前炎症小体信号通路中研究最为核心的胞质内受体,可感受 PAMPs、DAMPs 及多种疾病密切关联的分子(如 AD 中的 Aβ)活化。故 NLRP3 炎症小体在神经退行性疾病等多种疾病中均发挥着至关重要的作用。
本研究证明了在 tau 蛋白引发的神经退行性疾病中,NLRP3 炎症小体的激活是驱动 tau 蛋白过磷酸化的重要因素。
NLRP3 炎症小体的激活和炎症调控机制的阐明,填补了 AD 炎症发病学说的关键环节,揭示了炎症过程促进 AD 等神经退行性脑疾病的机制,同时理清了 AD 三大核心要素(Aβ 沉积、tau 蛋白过磷酸化和神经炎症)的联系。这一证据毫无疑问解开了炎症反应是否能驱动及如何驱动 AD 发生的疑问。
最后,对于 AD 患者面对的无药可医困境,NLRP3 炎症小体为阿尔茨海默病药物研发提供新的思路。

图片来源:CISION
值得欣喜的是,已有 NLRP3 拮抗剂类药物进入临床试验阶段。在这篇研究的作者单位中,除了研究机构外,我们同样看到了企业的身影。
NLRP3 因可被多种信号激活,与多种疾病(老年退行性病变、免疫性疾病和肿瘤)的发生发展有关。诺华在今年四月以 12.6 亿美元收购了开发 NLRP3 拮抗剂的子公司 IFM Tre,后者的核心技术产品是已经进入临床的 NLRP3 拮抗剂 IFM- 2427。
这种良性的资本驱动以及企业和研究机构科研人员的合作可以极大的推动研究成果转化。
延伸阅读
2019 年 9 月 11 日,来自美国圣犹达儿童研究医院等研究机构的研究人员在 Nature 上发表题为《DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome》的研究成果,鉴定出一种称为 DDX3X 的分子,能通过调节 NLRP3 炎症小体决定着应激细胞的生死。

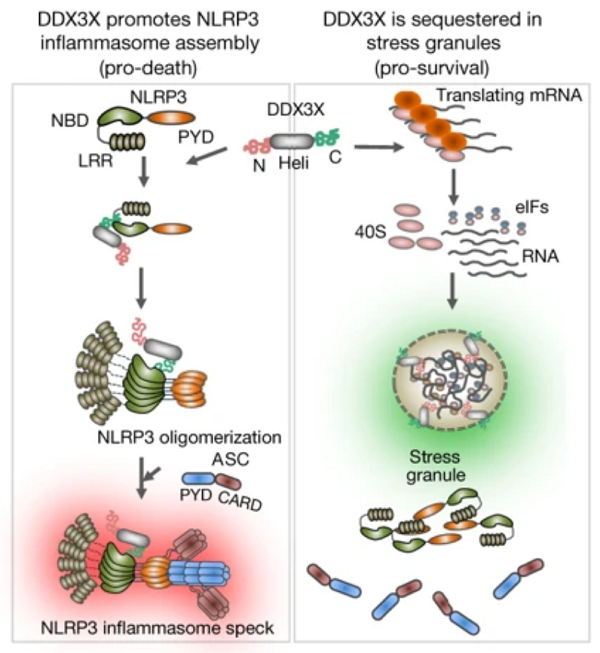
图片来源:nature
细胞遭受应激时会形成应激颗粒。应激颗粒的形成和 NLRP3 炎症小体激活对 DDX3X(一种新鉴定的分子,在决定遭受应激的细胞的命运方面起着关键作用)的竞争使得巨噬细胞能够理解应激信号并选择它们的命运。
应激颗粒的形成特异性地抑制了 DDX3X 对激活 NLRP3 炎症小体的可用性,抑制细胞焦亡这种细胞死亡途径。
本研究为自身炎症疾病和其他疾病提供了新的可能靶点。
2019 年 6 月 12 日,北京大学毛有东课题组与哈佛医学院吴皓课题组合作在 Nature 杂志发表题为《Structural mechanism for NEK7 -licensed activation of NLRP3 inflammasome》的研究长文,系统揭示了 NEK7 与 NLRP3 亚基的多界面相互作用,以及其介导 NLRP3 炎症小体激活的分子机制。

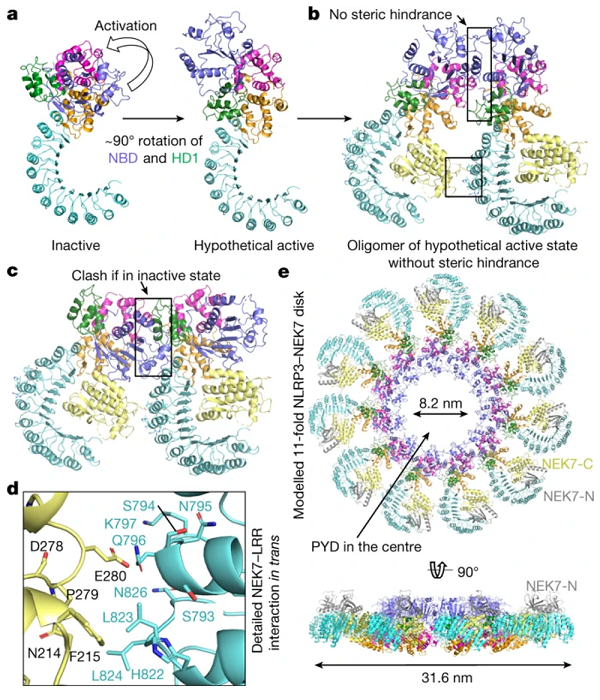
图片来源:nature
NEK7 在有丝分裂间期中参与组装和激活 NLRP3 炎症小体。
对 NLRP3 的结构和功能的研究证明 NLRP3 -NEK7 相互作用决定了 NEK7 许可对 NLRP3 炎症小体激活的必要性,为进一步研究 NLRP3 炎症小体激活的分子机制提供了重要基础,为相关疾病的分子医学研究提供了必备的分子依据和药物治疗靶点。